Clear Sky Science · ja
単一細胞RNAおよびT細胞受容体シーケンスデータの共同解析のためのマルチモーダルフレームワークはがん免疫療法に対するT細胞応答を予測する
免疫応答を予測することが重要な理由
チェックポイント阻害薬のようながん免疫療法は、患者自身のT細胞を解き放ち腫瘍を攻撃させることで効果を発揮します。しかし、恩恵を受ける患者は限られており、医師は誰が反応するかを予測する手段が乏しいのが現状です。本研究はTRIMと呼ばれる計算フレームワークを提示します。TRIMは血液や腫瘍中のT細胞を詳細に単一細胞レベルで計測したデータから学習します。各細胞の遺伝子発現と固有のT細胞受容体に関する情報を組み合わせることで、治療後にT細胞がどのように増殖・変化するかを予測し、日常的な血液検査から治療成功を見通す手段を提供する可能性があります。
個々のT細胞を覗く
T細胞は体内を巡回し、特定の分子断片を認識するT細胞受容体を使って危険の兆候を探します。がんに遭遇すると、あるT細胞はほぼ同一のクローンへと増殖し、能動的な腫瘍殺傷者から効果を失った疲弊細胞までさまざまな役割をとることがあります。現在の単一細胞技術では、各T細胞について遺伝子発現プロファイル(どの遺伝子がオン・オフか)と受容体配列の両方を読み取ることが可能です。著者らはこの手法を、頭頸部がん、大腸がん、ほか複数のがん患者から得た数千個のT細胞に適用し、免疫療法の前後で血液と腫瘍組織の双方をサンプリングしました。
複雑な計測から共有マップへ
TRIMは条件付き変分オートエンコーダとして知られる深層学習アーキテクチャに基づいています。簡単に言えば、各細胞の遺伝子と受容体から得られる豊富な情報を共有の低次元マップへ圧縮し、同時に患者情報、細胞の由来(血液か腫瘍か)、採取時点(治療前か後か)も考慮します。遺伝子活動と受容体特徴は別々の入力モジュールで読み取られ、別々の出力モジュールで再構築を試みます。特殊な損失関数により、同一クローンに属する細胞はこのマップ上で近くに配置され、異なるクローンは離れるように促されます。この設計により、モデルは生物学的に最も重要な点、すなわち各クローンの大きさとその細胞が機能的な状態にどのように分布しているかに焦点を当てます。
受容体、遺伝子活動、クローンを結ぶもの
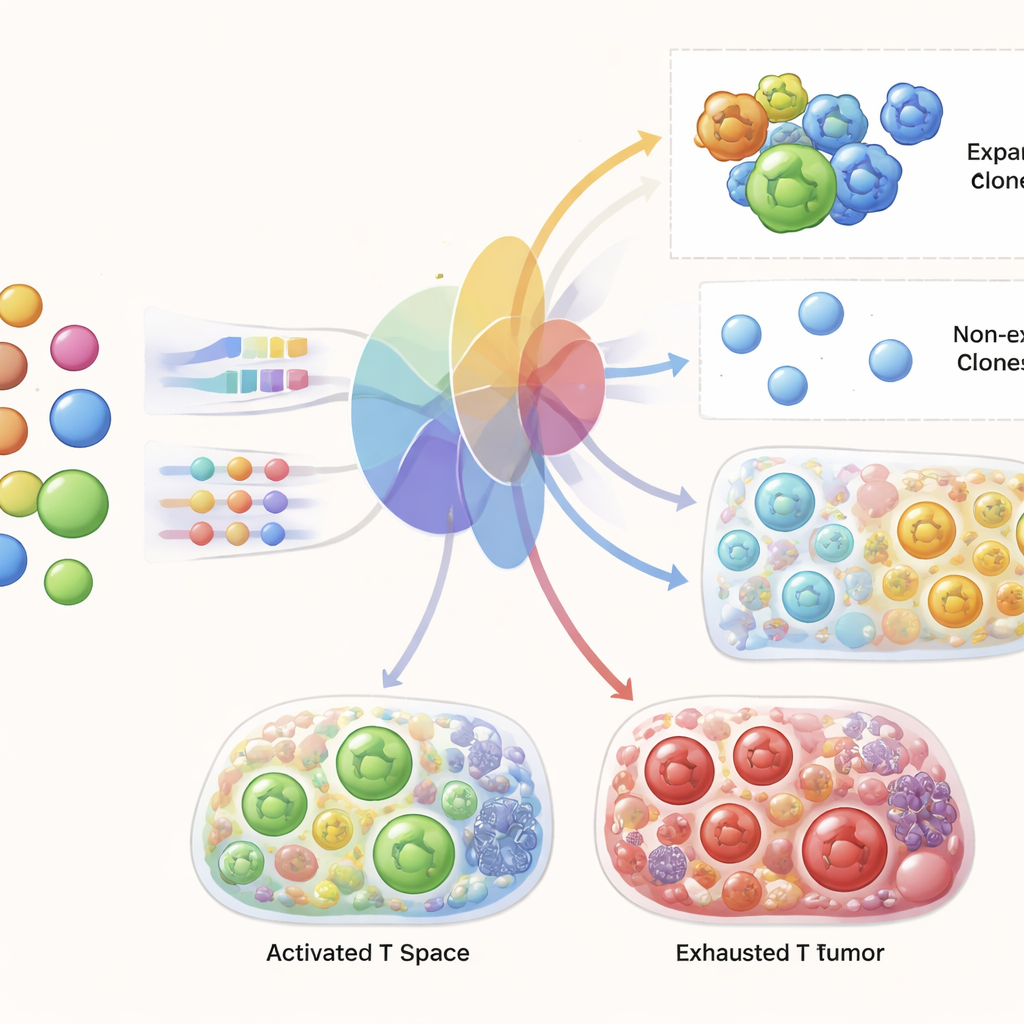
複数のデータセットにわたり受容体類似性と遺伝子発現を体系的に比較した結果、まったく同一の受容体を共有する細胞は、ランダムなT細胞に比べ遺伝子発現が確かにより類似していることが分かりました。とくに大きく拡張したクローンでその傾向が顕著でした。しかし、完全一致を超えると、受容体配列の小さな変化は遺伝子発現の類似性と一貫して対応するわけではありません。言い換えれば、ほとんど同一の受容体を持つ細胞が、まったく無関係な受容体を持つ細胞と同程度に異なる表現型を示すことがあります。これにより研究チームは、受容体情報と細胞状態の間の重要な橋渡しとして、細かい配列類似性よりもクローンの同一性とクローンサイズに注目しました。
血液から腫瘍結合T細胞を予測する
TRIMの中心的な試験は、患者の治療前血液データのみを用いて、その患者の治療後の腫瘍および血液中のT細胞を現実的に予測できるかどうかでした。著者らは1人の患者を除いてTRIMを学習させ、除いた患者を予測させることをコホートごとに繰り返しました。モデルは条件間でのT細胞遺伝子状態とクローン多様性の全体像を正確に捉えていることを示しました。さらに注目すべきは、治療前に存在した個々の受容体クローンについて、TRIMが治療後にどのクローンが拡大するかを高い精度で予測でき、標準的な機械学習手法を明確に上回った点です。また、活性化し増殖するCD8 T細胞の増加や効果的な腫瘍殺傷に関連する特定の遺伝子プログラムの増強など、既知の成功応答シグネチャも再現しました。
成功する拡張の背後にある遺伝子を明らかにする
TRIMは内部でどの細胞が拡大するかを判断する必要があるため、研究者はモデルを解析してどの遺伝子に依拠しているかを調べることができました。TRIMが指摘した遺伝子は、T細胞活性化の中核的特徴と一致します:増殖を制御する細胞周期調節因子、標的細胞を殺す分子、活性化とコミュニケーションを支えるタンパク質、さらに腫瘍内への移動や機能を助ける代謝・移動関連因子です。重要なことに、モデルは予測時に治療前の血液データのみを用いてこれらのプログラムを同定しており、循環するT細胞の初期の分子差異が、治療開始後に腫瘍のシグナルに遭遇した際の応答を予見していることを示唆します。
患者にとっての意義
要するに、この研究は慎重に設計されたモデルがマルチモーダルな単一細胞データから学び、がん免疫療法中に患者のT細胞がどのように振る舞うかを予測できることを示しています。TRIMは治療前の血液サンプルを用いて、どのクローンが拡大しどの状態に入るか、そしてどの遺伝子プログラムがその振る舞いを駆動するかを、直接採取が困難な組織においても推定します。臨床利用にはさらなる検証と大規模データが必要ですが、このアプローチは、TRIMのようなモデルと簡便な血液検査を組み合わせて治療反応を予測し、病勢を監視し、個別化免疫療法の指針となる新たなバイオマーカーを発見する未来を示唆しています。
引用: He, C., Amodio, M., Ashenberg, O. et al. Multimodal framework for the joint analysis of single-cell RNA and T cell receptor sequencing data predicts T cell response to cancer immunotherapy. Nat Commun 17, 3840 (2026). https://doi.org/10.1038/s41467-026-70505-0
キーワード: がん免疫療法, T細胞, 単一細胞シーケンシング, 機械学習モデル, 腫瘍バイオマーカー