Clear Sky Science · fr
Cadre multimodal pour l’analyse conjointe des données de séquençage ARN unicellulaire et des récepteurs des cellules T prédit la réponse des cellules T à l’immunothérapie du cancer
Pourquoi il est important de prédire la réponse immunitaire
Les immunothérapies contre le cancer, comme les inhibiteurs de points de contrôle, agissent en libérant les cellules T du patient pour qu’elles attaquent les tumeurs. Pourtant, seuls certains patients en bénéficient, et les médecins disposent aujourd’hui d’outils limités pour prévoir qui répondra. Cette étude présente un cadre computationnel appelé TRIM qui apprend à partir de mesures unicellulaires détaillées des cellules T dans le sang et les tumeurs. En combinant l’information sur l’activité génique de chaque cellule et sa séquence unique de récepteur des cellules T, TRIM peut prédire comment les cellules T vont se développer et évoluer après le traitement, offrant un moyen potentiel d’anticiper le succès d’une thérapie à partir d’une prise de sang de routine.
Observer l’intérieur des cellules T individuelles
Les cellules T patrouillent l’organisme, à la recherche de signes de danger grâce à leurs récepteurs, qui reconnaissent des fragments moléculaires spécifiques. Lorsqu’une cellule T rencontre un cancer, elle peut se multiplier en un clone composé de nombreuses cellules presque identiques et adopter différents rôles, des effectrices actives anti-tumorales aux cellules épuisées incapables de lutter efficacement. Les technologies unicellulaires modernes peuvent désormais lire, pour chaque cellule T, à la fois son profil d’expression génique (quels gènes sont activés ou désactivés) et la séquence de son récepteur. Les auteurs ont appliqué ces outils à des milliers de cellules T issues de patients atteints de cancers de la tête et du cou, colorectaux et d’autres types, en échantillonnant à la fois le sang et le tissu tumoral avant et après immunothérapie.
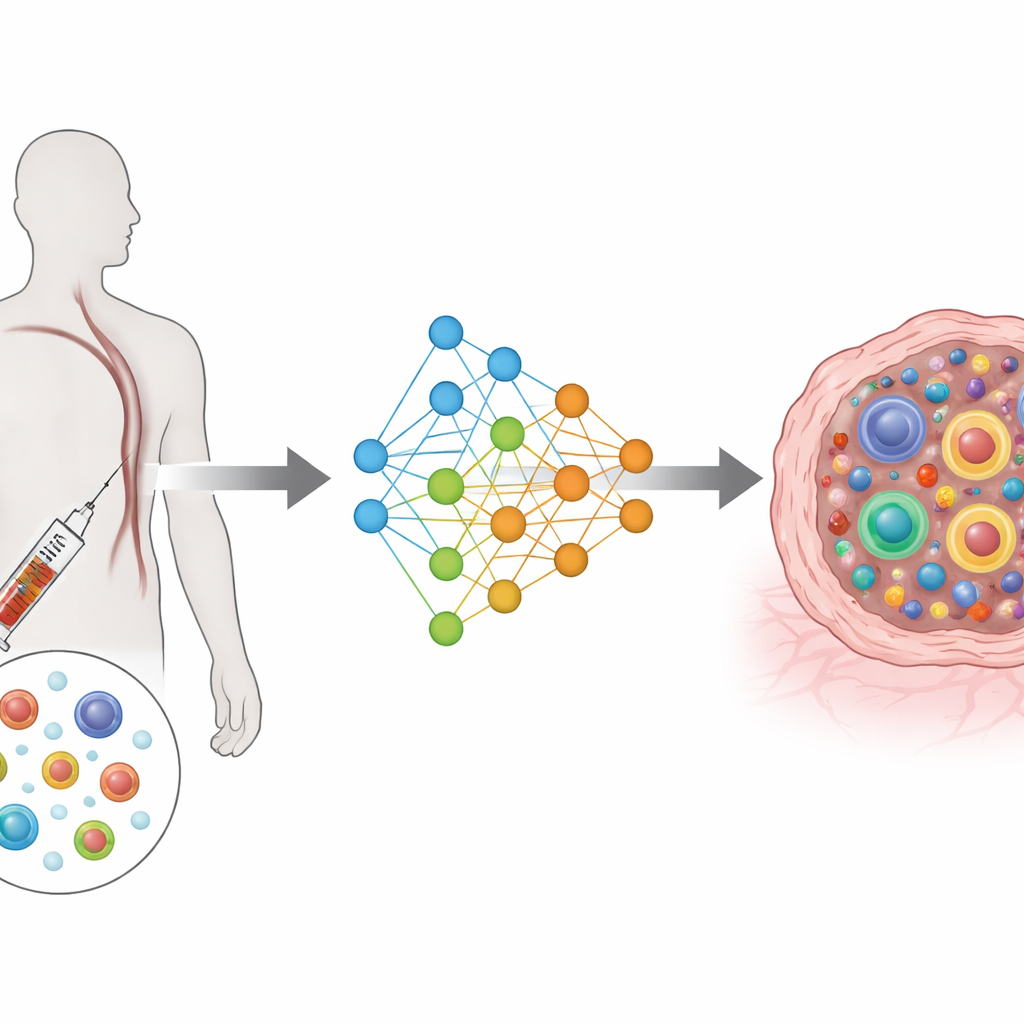
Des mesures complexes à une carte partagée
TRIM repose sur un type d’architecture d’apprentissage profond connu sous le nom d’autoencodeur variationnel conditionnel. En termes simples, il compresse les informations riches provenant des gènes et du récepteur de chaque cellule en une carte partagée de faible dimension, tout en tenant compte du patient, de l’origine de la cellule (sang ou tumeur) et du moment de prélèvement (avant ou après traitement). Des modules d’entrée séparés lisent l’activité génique et les caractéristiques du récepteur, et des modules de sortie séparés tentent de les reconstruire. Une fonction de perte spéciale encourage les cellules appartenant au même clone à se regrouper dans cette carte, tout en maintenant une séparation entre les clones différents. Cette conception concentre le modèle sur ce qui importe biologiquement : la taille de chaque clone et la répartition de ses cellules selon des états fonctionnels.
Ce qui relie récepteurs, activité génique et clones
En comparant systématiquement la similarité des récepteurs et l’expression génique à travers plusieurs jeux de données, les chercheurs ont constaté que les cellules partageant exactement le même récepteur sont effectivement plus similaires entre elles en termes d’expression génique que des cellules T prises au hasard, en particulier pour les clones larges et étendus. Cependant, au-delà des correspondances exactes, de petits changements dans la séquence du récepteur ne prédisent pas de manière fiable la similarité d’expression génique entre deux cellules. Autrement dit, des cellules ayant des récepteurs presque identiques peuvent être aussi différentes que des cellules aux récepteurs complètement distincts. Cela a conduit l’équipe à se concentrer sur l’identité du clone et la taille du clone, plutôt que sur la similarité fine des séquences de récepteur, comme lien essentiel entre l’information du récepteur et l’état cellulaire.
Prédire les cellules T présentes dans la tumeur à partir du sang
Le test central pour TRIM était de savoir s’il pouvait utiliser uniquement les données sanguines pré-traitement d’un patient pour générer des prédictions réalistes pour les cellules T de ce patient dans la tumeur et dans le sang après la thérapie. Les auteurs ont entraîné TRIM sur tous les patients sauf un, puis lui ont demandé de prédire le patient mis de côté, en répétant cette procédure à travers les cohortes. Ils ont montré que le modèle capture avec précision le paysage global des états géniques des cellules T et la diversité des clones selon les conditions. Plus frappant, pour des clones de récepteurs individuels présents avant la thérapie, TRIM a pu prédire avec une grande précision lesquels s’étendraient après le traitement, surpassant nettement les approches classiques d’apprentissage automatique. Il a également reproduit des signatures connues de réponse réussie, telles qu’une augmentation des cellules CD8 activées et en prolifération et des programmes géniques spécifiques liés à une élimination tumorale efficace.
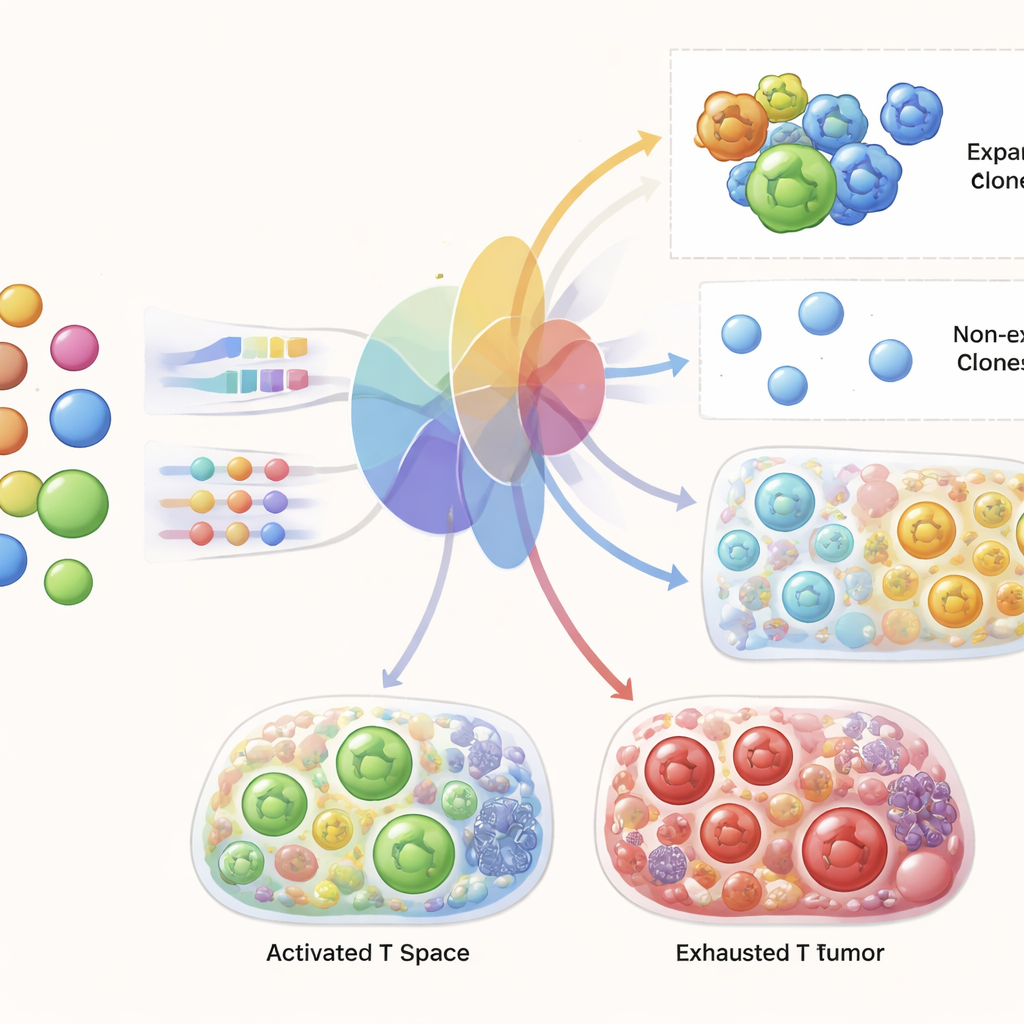
Mettre au jour les gènes derrière une expansion réussie
Parce que TRIM doit déterminer en interne quelles cellules sont susceptibles de s’étendre, les auteurs ont pu sonder le modèle pour identifier les gènes sur lesquels il s’appuie. Les gènes mis en évidence par TRIM correspondent aux caractéristiques centrales de l’activation des cellules T : régulateurs du cycle cellulaire contrôlant la prolifération, molécules impliquées dans la destruction des cibles, protéines gouvernant l’activation et la communication, ainsi que des facteurs métaboliques et de migration aidant les cellules T à pénétrer et fonctionner dans les tumeurs. Fait important, le modèle a identifié ces programmes en n’utilisant que des données sanguines pré-traitement au moment de la prédiction, ce qui suggère que des différences moléculaires précoces chez les cellules T circulantes préfigurent leur manière de répondre une fois la thérapie lancée et que ces cellules rencontrent les signaux tumoraux.
Qu’est-ce que cela implique pour les patients
En substance, ce travail montre qu’un modèle soigneusement conçu peut apprendre à partir de données unicellulaires multimodales pour prévoir le comportement des cellules T d’un patient pendant une immunothérapie du cancer. TRIM utilise des échantillons sanguins pré-traitement pour inférer quels clones s’étendront, quels états ils adopteront et quels programmes géniques piloteront ce comportement, y compris dans des tissus difficiles à échantillonner directement. Bien que des validations supplémentaires et des jeux de données plus larges soient nécessaires avant un usage clinique, l’approche ouvre la voie à un futur où les oncologues pourraient utiliser un simple test sanguin, associé à des modèles comme TRIM, pour prédire la réponse au traitement, surveiller la progression de la maladie et découvrir de nouveaux biomarqueurs guidant des décisions d’immunothérapie personnalisée.
Citation: He, C., Amodio, M., Ashenberg, O. et al. Multimodal framework for the joint analysis of single-cell RNA and T cell receptor sequencing data predicts T cell response to cancer immunotherapy. Nat Commun 17, 3840 (2026). https://doi.org/10.1038/s41467-026-70505-0
Mots-clés: immunothérapie du cancer, cellules T, séquençage unicellulaire, modèle d’apprentissage automatique, biomarqueurs tumoraux