Clear Sky Science · nl
Multimodaal kader voor gezamenlijke analyse van single-cell RNA- en T-celreceptor-sequencinggegevens voorspelt T-celrespons op kankerimmunotherapie
Waarom het voorspellen van immuunrespons belangrijk is
Kankerimmunotherapieën, zoals remmers van immuuncheckpoints, werken door de eigen T-cellen van een patiënt vrij te maken om tumoren aan te vallen. Toch profiteert maar een deel van de patiënten, en artsen hebben momenteel beperkte middelen om te voorspellen wie zal reageren. Deze studie introduceert een computationeel kader genaamd TRIM dat leert van gedetailleerde single-cell metingen van T-cellen in bloed en tumoren. Door informatie over de genactiviteit van elke cel te combineren met diens unieke T-celreceptor, kan TRIM voorspellen hoe T-cellen zich zullen uitbreiden en veranderen na behandeling, wat een manier biedt om het behandelingssucces te anticiperen aan de hand van een routinematige bloedafname.
Inzicht in individuele T-cellen
T-cellen patrouilleren door het lichaam en zoeken naar tekenen van gevaar met hun T-celreceptoren, die specifieke moleculaire fragmenten herkennen. Wanneer een T-cel kanker tegenkomt, kan deze zich vermenigvuldigen tot een kloon van veel vrijwel identieke cellen en verschillende rollen aannemen, van actieve tumorverdelgers tot uitgeputte cellen die niet langer effectief kunnen vechten. Moderne single-cell technologieën kunnen nu per T-cel zowel het genexpressieprofiel (welke genen aan- of uitgezet zijn) als de receptorsequentie uitlezen. De auteurs pasten deze instrumenten toe op duizenden T-cellen van patiënten met hoofd-hals-, colorectale en verschillende andere kankers, met monsters van zowel bloed als tumorweefsel vóór en na immunotherapie.
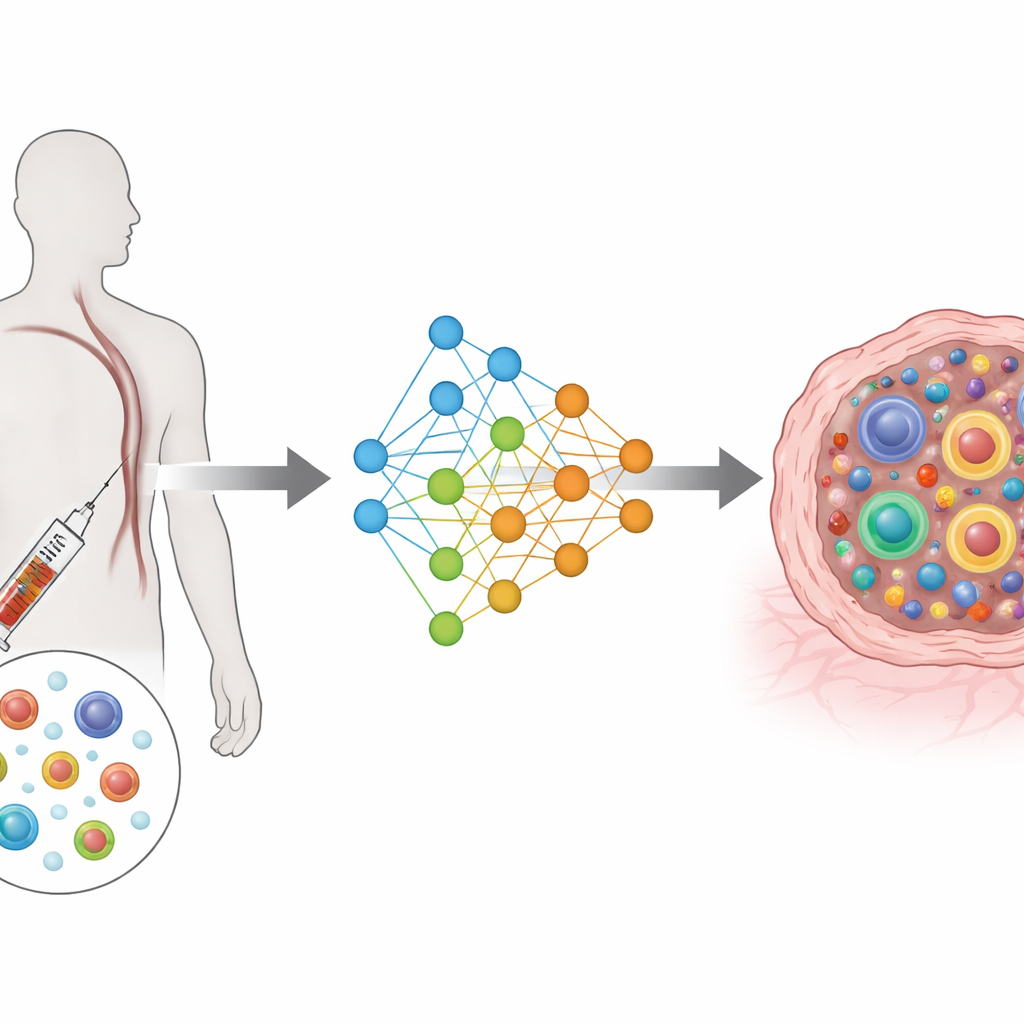
Van complexe metingen naar een gedeelde kaart
TRIM is gebouwd op een type deep learning-architectuur dat bekendstaat als een conditionele variational autoencoder. In eenvoudige termen comprimeert het de rijke informatie van de genen en receptor van elke cel tot een gedeelde laag-dimensionale kaart, terwijl het ook rekening houdt met wie de patiënt is, waar de cel vandaan kwam (bloed of tumor) en wanneer het monster is genomen (voor of na behandeling). Afzonderlijke inputmodules lezen de genactiviteit en receptorfeatures, en afzonderlijke outputmodules proberen deze te reconstrueren. Een speciale verliesfunctie zorgt ervoor dat cellen die tot dezelfde kloon behoren dicht bij elkaar komen te liggen in deze kaart, terwijl verschillende klonen uit elkaar gehouden worden. Dit ontwerp richt het model op wat biologisch het belangrijkst is: hoe groot elke kloon is en hoe de cellen daarvan verdeeld zijn over functionele toestanden.
Wat receptoren, genactiviteit en klonen verbindt
Door receptorovereenkomst en genexpressie systematisch te vergelijken over meerdere datasets, vonden de onderzoekers dat cellen die exact dezelfde receptor delen inderdaad meer op elkaar lijken in genactiviteit dan willekeurige T-cellen, vooral voor grote, uitgebreide klonen. Echter, buiten exacte overeenkomsten volgen kleine veranderingen in de receptorsequentie niet betrouwbaar hoe vergelijkbaar twee cellen zijn in hun genexpressie. Met andere woorden: cellen met bijna identieke receptoren kunnen er net zo verschillend uitzien als cellen met volledig niet-verwante receptoren. Dit bracht het team ertoe zich te concentreren op kloonidentiteit en kloongrootte, in plaats van fijnmazige receptorsequentie-overeenkomst, als de cruciale schakel tussen receptorinformatie en celtoestand.
Voorspellen van tumorgebonden T-cellen vanuit bloed
De centrale toets voor TRIM was of het alleen met pre-behandelingsbloedgegevens van een patiënt realistische voorspellingen kon maken voor die patiënts T-cellen in de tumor en in het bloed na therapie. De auteurs trainden TRIM op alle patiënten behalve één en vroegen het model vervolgens de weggelaten patiënt te voorspellen, en herhaalden dit over cohorten. Ze toonden aan dat het model het algemene landschap van T-cel genstaten en kloondiversiteit over condities nauwkeurig vastlegt. Nog opvallender, voor individuele receptorklonen die vóór therapie aanwezig waren, kon TRIM met hoge nauwkeurigheid voorspellen welke zouden expanderen na behandeling, en overtrof het duidelijk standaard machine-learningmethoden. Het reproduceerde ook bekende signaturen van succesvolle respons, zoals een toename van geactiveerde, delende CD8 T-cellen en specifieke genprogramma's die gekoppeld zijn aan effectieve tumorverdeling.
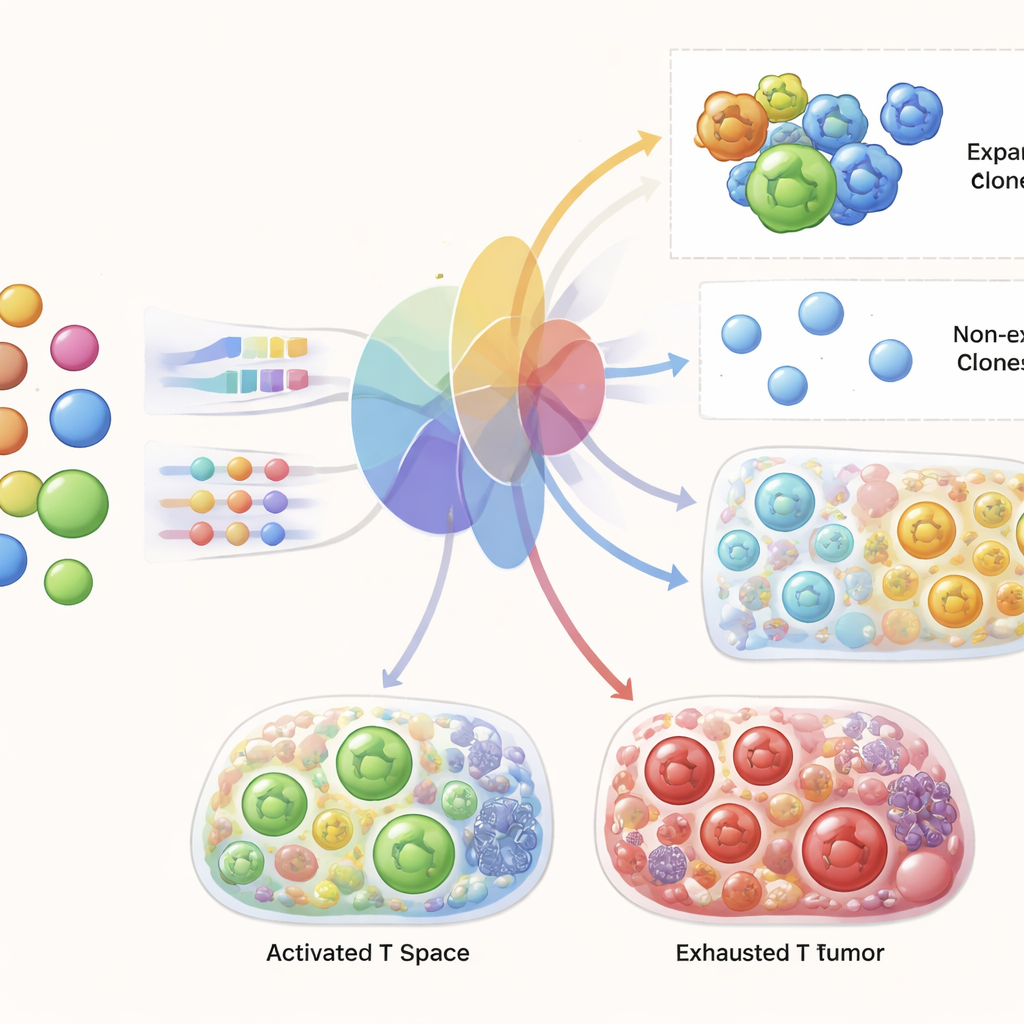
De genen achter succesvolle expansie onthullen
Aangezien TRIM intern moet bepalen welke cellen waarschijnlijk zullen uitbreiden, konden de auteurs het model onderzoeken om te zien op welke genen het vertrouwt. De door TRIM gemarkeerde genen komen overeen met kernkenmerken van T-celactivatie: celcyclusregulatoren die proliferatie sturen, moleculen betrokken bij het doden van doelcellen, eiwitten die activatie en communicatie regelen, en metabolische en migratie-gerelateerde factoren die T-cellen helpen tumorweefsel binnen te dringen en daar te functioneren. Belangrijk is dat het model deze programma's identificeerde met alleen pre-behandelingsbloedgegevens op het moment van voorspelling, wat suggereert dat vroege moleculaire verschillen in circulerende T-cellen voorspellen hoe ze zullen reageren zodra de therapie begint en ze tumorsignalen tegenkomen.
Wat dit betekent voor patiënten
In wezen toont dit werk aan dat een zorgvuldig ontworpen model kan leren van multimodale single-cellgegevens om te voorspellen hoe de T-cellen van een patiënt zich zullen gedragen tijdens kankerimmunotherapie. TRIM gebruikt pre-behandelingsbloedmonsters om af te leiden welke klonen zullen uitbreiden, in welke toestanden ze terechtkomen en welke genprogramma's dit gedrag aansturen, zelfs in weefsels die moeilijk direct te bemonsteren zijn. Hoewel verdere validatie en grotere datasets nodig zijn voordat klinisch gebruik mogelijk is, wijst de benadering op een toekomst waarin oncologen met een eenvoudige bloedtest, gecombineerd met modellen zoals TRIM, behandelingsrespons kunnen voorspellen, ziekteprogressie kunnen monitoren en nieuwe biomarkers kunnen ontdekken die gepersonaliseerde immunotherapie sturen.
Bronvermelding: He, C., Amodio, M., Ashenberg, O. et al. Multimodal framework for the joint analysis of single-cell RNA and T cell receptor sequencing data predicts T cell response to cancer immunotherapy. Nat Commun 17, 3840 (2026). https://doi.org/10.1038/s41467-026-70505-0
Trefwoorden: kankerimmunotherapie, T-cellen, single-cell sequencing, machine learning-model, tumormarkers