Clear Sky Science · pl
Wielomodalne ramy do wspólnej analizy sekwencjonowania RNA pojedynczych komórek i receptorów T — przewidywanie reakcji komórek T na immunoterapię przeciwnowotworową
Dlaczego prognozowanie odpowiedzi układu odpornościowego ma znaczenie
Immunoterapie przeciwnowotworowe, takie jak inhibitory punktów kontrolnych, działają przez uruchomienie własnych komórek T pacjenta do ataku na guzy. Jednak korzysta z nich tylko część chorych, a lekarze mają obecnie ograniczone narzędzia do przewidywania, kto zareaguje. W tym badaniu przedstawiono ramy obliczeniowe o nazwie TRIM, które uczą się na podstawie szczegółowych pomiarów pojedynczych komórek T we krwi i w tkankach nowotworowych. Łącząc informacje o aktywności genów w poszczególnych komórkach oraz o ich unikatowych receptorach T, TRIM potrafi przewidzieć, które komórki T będą się rozszerzać i jak się zmienią po leczeniu, oferując potencjalny sposób na przewidzenie powodzenia terapii na podstawie rutynowego badania krwi.
Zajrzeć do wnętrza pojedynczych komórek T
Komórki T patrolują organizm, poszukując oznak zagrożenia za pomocą receptorów T, które rozpoznają określone fragmenty molekularne. Gdy komórka T natrafi na nowotwór, może się rozmnożyć do klonu wielu niemal identycznych komórek i przyjmować różne role — od aktywnych zabójców guza po wyczerpane komórki, które już nie walczą skutecznie. Nowoczesne technologie pojedynczych komórek pozwalają dziś odczytać dla każdej komórki zarówno profil ekspresji genów (które geny są włączone lub wyłączone), jak i sekwencję jej receptora. Autorzy zastosowali te narzędzia do tysięcy komórek T od pacjentów z rakiem głowy i szyi, jelita grubego oraz innymi nowotworami, pobierając próbki zarówno z krwi, jak i z tkanki nowotworowej przed i po immunoterapii.
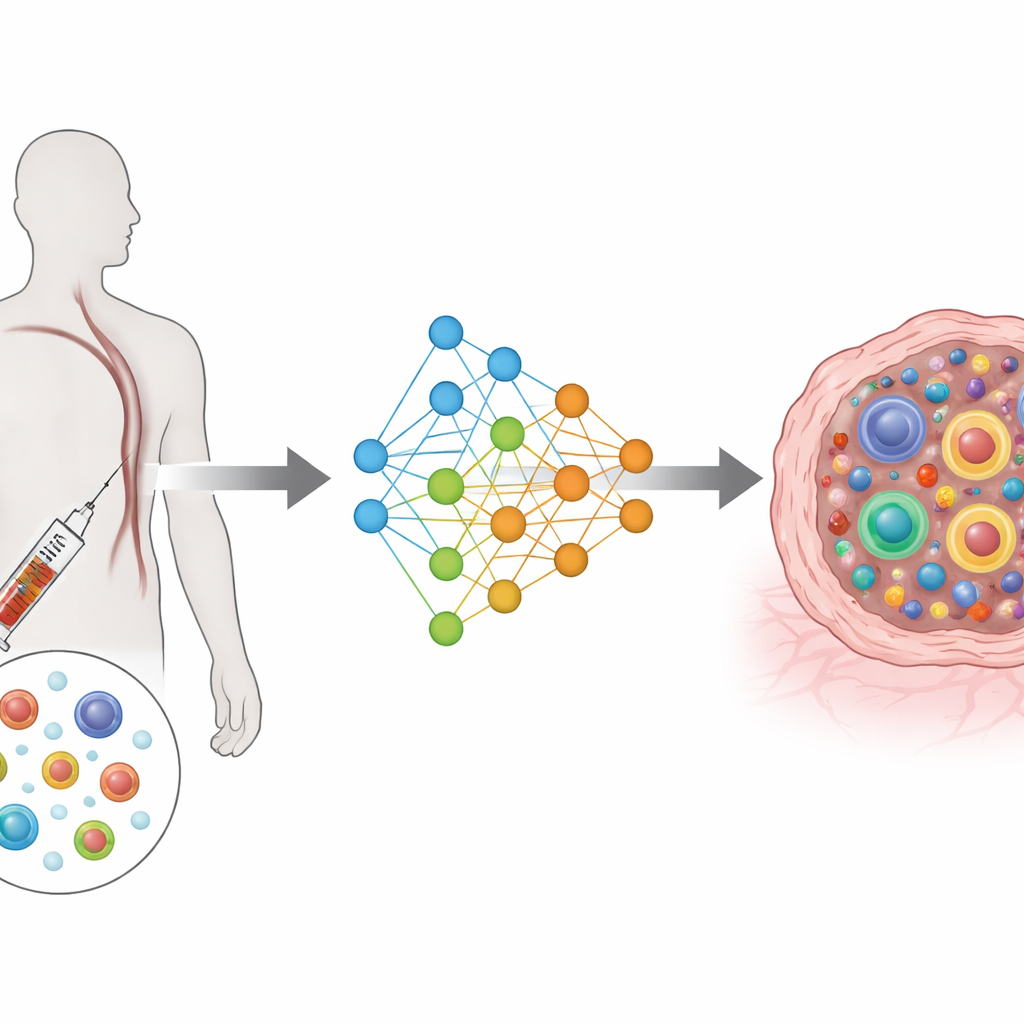
Od złożonych pomiarów do wspólnej mapy
TRIM opiera się na typie architektury głębokiego uczenia znanej jako warunkowy autoenkoder wariacyjny. Mówiąc prosto, kompresuje bogate informacje z genów i receptorów każdej komórki do wspólnej, niskowymiarowej mapy, uwzględniając przy tym, kim jest pacjent, skąd pochodziła komórka (krew czy guz) oraz kiedy została pobrana (przed lub po leczeniu). Osobne moduły wejściowe odczytują aktywność genów i cechy receptorów, a osobne moduły wyjściowe próbują je odtworzyć. Specjalna funkcja straty skłania komórki należące do tego samego klonu do zajmowania pobliskich miejsc na tej mapie, jednocześnie utrzymując różne klony odseparowane. Taka konstrukcja koncentruje model na tym, co biologicznie najważniejsze: na wielkości każdego klonu i na tym, jak jego komórki rozkładają się między stanami funkcjonalnymi.
Co łączy receptory, aktywność genów i klony
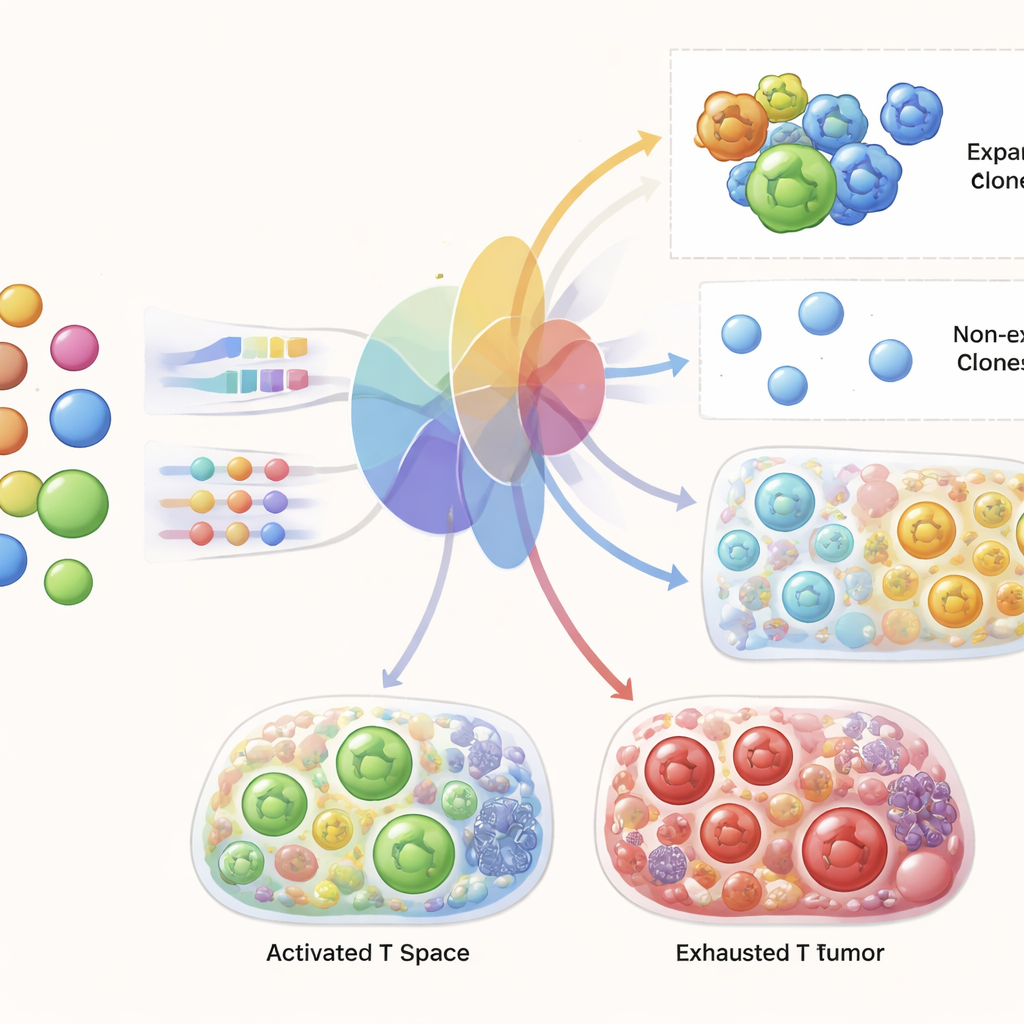
Poprzez systematyczne porównanie podobieństwa receptorów i ekspresji genów w wielu zestawach danych, badacze stwierdzili, że komórki dzielące dokładnie ten sam receptor rzeczywiście są bardziej podobne pod względem ekspresji genów niż losowe komórki T, szczególnie w przypadku dużych, rozszerzonych klonów. Jednak poza dokładnymi dopasowaniami drobne zmiany w sekwencji receptora nie korelują wiarygodnie z podobieństwem w ekspresji genów. Innymi słowy, komórki o niemal identycznych receptorach mogą wyglądać tak różnie, jak komórki o zupełnie niepowiązanych receptorach. To skłoniło zespół do skupienia się na tożsamości klonu i jego wielkości, zamiast na drobiazgowej podobieństwie sekwencji receptora, jako kluczowego mostu między informacją o receptorze a stanem komórki.
Przewidywanie komórek T w guzie na podstawie krwi
Głównym testem dla TRIM było sprawdzenie, czy model potrafi użyć wyłącznie danych z krwi pobranej przed leczeniem od pacjenta i wygenerować realistyczne przewidywania dotyczące komórek T tego pacjenta w guzie i we krwi po terapii. Autorzy trenowali TRIM na wszystkich pacjentach poza jednym, a następnie prosili go o przewidzenie pacjenta wyłączonego z treningu, powtarzając to w ramach kohort. Pokazali, że model trafnie odwzorowuje ogólny krajobraz stanów genowych komórek T i różnorodność klonów w różnych warunkach. Co bardziej imponujące, dla poszczególnych klonów receptorowych obecnych przed terapią TRIM potrafił z dużą dokładnością przewidzieć, które z nich rozszerzą się po leczeniu, zdecydowanie przewyższając standardowe podejścia uczenia maszynowego. Model odtworzył też znane sygnatury skutecznej odpowiedzi, takie jak wzrost aktywowanych, proliferujących komórek CD8 i specyficznych programów genowych związanych ze skutecznym zabijaniem nowotworu.
Odkrywanie genów stojących za udanym rozszerzeniem
Ponieważ TRIM musi wewnętrznie ocenić, które komórki prawdopodobnie się rozszerzą, autorzy mogli zbadać model, by zobaczyć, na których genach się opiera. Geny wskazywane przez TRIM pokrywają się z podstawowymi cechami aktywacji komórek T: regulatorami cyklu komórkowego kontrolującymi proliferację, cząsteczkami zaangażowanymi w zabijanie komórek docelowych, białkami sterującymi aktywacją i komunikacją oraz czynnikami metabolicznymi i związanymi z migracją, które pomagają komórkom T przemieszczać się do guza i funkcjonować w jego środowisku. Co istotne, model zidentyfikował te programy używając jedynie danych z krwi pobranej przed leczeniem w czasie przewidywania, co sugeruje, że wczesne różnice molekularne w krążących komórkach T zapowiadają, jak zareagują, gdy terapia się rozpocznie i napotkają sygnały z guza.
Co to oznacza dla pacjentów
W istocie praca ta pokazuje, że starannie zaprojektowany model potrafi uczyć się z wielomodalnych danych pojedynczych komórek, aby prognozować, jak komórki T pacjenta będą się zachowywać podczas immunoterapii przeciwnowotworowej. TRIM wykorzystuje próbki krwi pobrane przed leczeniem, by wnioskować, które klony się rozszerzą, w jakie stany wejdą i które programy genowe napędzają to zachowanie, nawet w tkankach trudnych do bezpośredniego pobrania. Chociaż przed zastosowaniem klinicznym potrzebna jest dalsza walidacja i większe zbiory danych, podejście to wskazuje na przyszłość, w której onkolodzy mogliby używać prostego badania krwi w połączeniu z modelami takimi jak TRIM do przewidywania odpowiedzi na terapię, monitorowania postępu choroby i odkrywania nowych markerów naprowadzających spersonalizowane decyzje terapeutyczne.
Cytowanie: He, C., Amodio, M., Ashenberg, O. et al. Multimodal framework for the joint analysis of single-cell RNA and T cell receptor sequencing data predicts T cell response to cancer immunotherapy. Nat Commun 17, 3840 (2026). https://doi.org/10.1038/s41467-026-70505-0
Słowa kluczowe: immunoterapia przeciwnowotworowa, komórki T, sekwencjonowanie pojedynczych komórek, model uczenia maszynowego, markery nowotworowe