Clear Sky Science · de
Multimodales Rahmenwerk zur gemeinsamen Analyse von Einzelzell‑RNA‑ und T‑Zell‑Rezeptor‑Sequenzierungsdaten sagt die T‑Zell‑Antwort auf Krebsimmuntherapie voraus
Warum die Vorhersage der Immunantwort wichtig ist
Krebsimmuntherapien, etwa Checkpoint‑Inhibitoren, wirken, indem sie die eigenen T‑Zellen eines Patienten befähigen, Tumoren anzugreifen. Dennoch profitieren nur einige Patienten, und Ärzte haben derzeit begrenzte Mittel, um vorherzusagen, wer ansprechen wird. Diese Studie stellt ein rechnerisches Rahmenwerk namens TRIM vor, das aus detaillierten Einzelzellmessungen von T‑Zellen im Blut und in Tumoren lernt. Indem es Informationen über die Genaktivität jeder Zelle und ihre einzigartige T‑Zell‑Rezeptorsequenz kombiniert, kann TRIM vorhersagen, wie sich T‑Zellen nach der Behandlung ausdehnen und verändern werden, und bietet so eine mögliche Methode, den Therapieerfolg aus einer routinemäßigen Blutprobe vorherzusehen.
Blick in einzelne T‑Zellen
T‑Zellen durchkämmen den Körper und suchen mit ihren T‑Zell‑Rezeptoren nach Gefahrenzeichen, indem sie spezifische molekulare Fragmente erkennen. Trifft eine T‑Zelle auf Krebs, kann sie sich zu einem Klon vieler nahezu identischer Zellen vermehren und unterschiedliche Rollen einnehmen, von aktiven Tumorzellen‑Killern bis zu erschöpften Zellen, die nicht mehr effektiv kämpfen können. Moderne Einzelzelltechnologien können nun für jede T‑Zelle sowohl das Genexpressionsprofil (welche Gene an- oder abgeschaltet sind) als auch die Rezeptorsequenz auslesen. Die Autoren wandten diese Werkzeuge auf Tausende von T‑Zellen von Patientinnen und Patienten mit Kopf‑Hals‑, Darm‑ und mehreren weiteren Krebsarten an und untersuchten Blut und Tumorgewebe vor und nach der Immuntherapie.
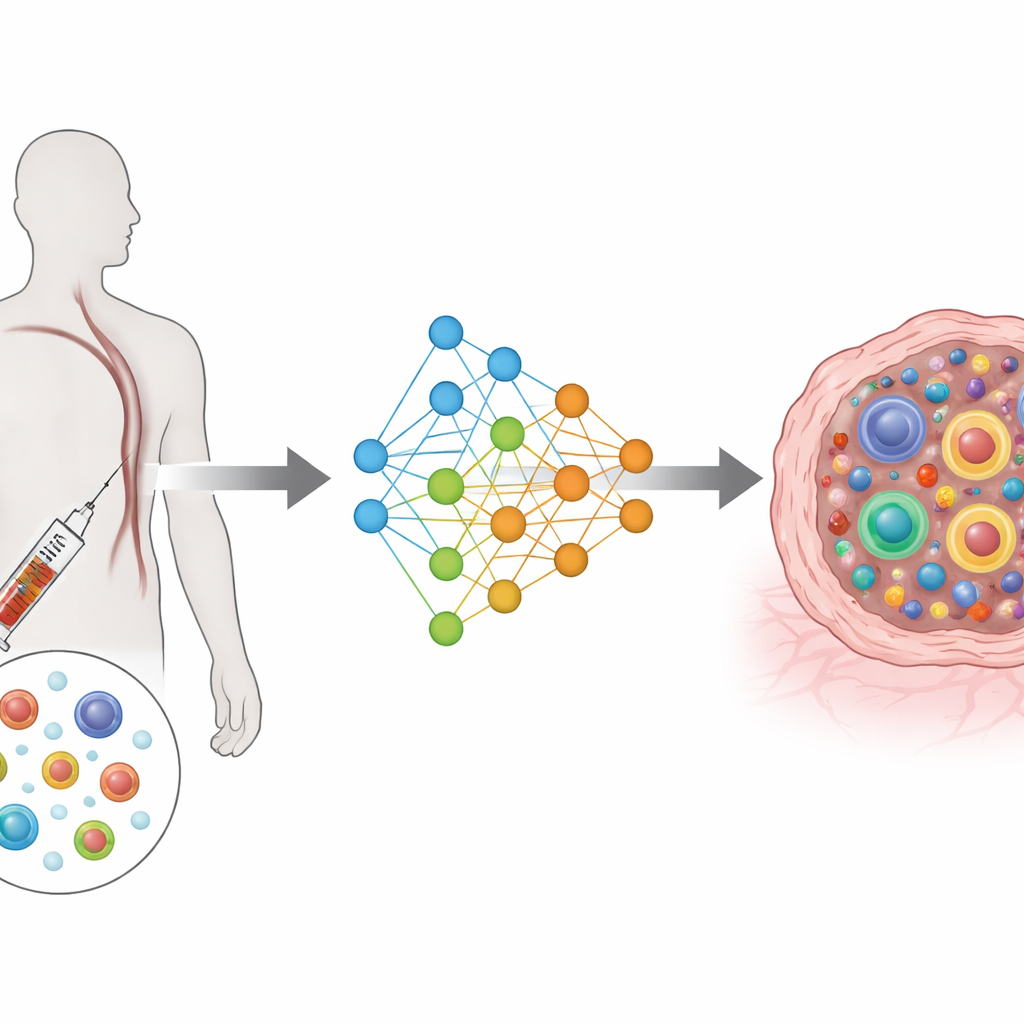
Von komplexen Messungen zu einer gemeinsamen Karte
TRIM basiert auf einer Art Deep‑Learning‑Architektur, die als bedingter variationaler Autoencoder bekannt ist. Vereinfacht gesagt komprimiert sie die reichhaltigen Informationen aus Genen und Rezeptoren jeder Zelle in eine gemeinsame, niedrigdimensionale Karte und berücksichtigt dabei auch, welcher Patient die Probe liefert, woher die Zelle stammt (Blut oder Tumor) und wann sie entnommen wurde (vor oder nach der Behandlung). Getrennte Eingabemodule lesen die Genaktivität und die Rezeptormerkmale, getrennte Ausgabemodule versuchen, diese wiederherzustellen. Eine spezielle Verlustfunktion bringt Zellen desselben Klons dazu, in dieser Karte nahe beieinander zu liegen, während verschiedene Klone voneinander getrennt bleiben. Dieses Design fokussiert das Modell auf das biologisch Wesentliche: die Größe jedes Klons und die Verteilung seiner Zellen auf funktionelle Zustände.
Was Rezeptoren, Genaktivität und Klone verbindet
Durch systematischen Vergleich von Rezeptorähnlichkeit und Genexpression über mehrere Datensätze fanden die Forschenden, dass Zellen mit genau demselben Rezeptor tatsächlich in ihrer Genaktivität ähnlicher sind als zufällige T‑Zellen, insbesondere bei großen, expandierten Klonen. Allerdings korreliert jenseits exakter Übereinstimmungen eine kleine Veränderung der Rezeptorsequenz nicht zuverlässig mit einer ähnlichen Genexpressionslage. Anders ausgedrückt: Zellen mit nahezu identischen Rezeptoren können ebenso unterschiedlich aussehen wie Zellen mit völlig unterschiedlichen Rezeptoren. Dies veranlasste das Team, sich auf Klonidentität und Klongröße statt auf fein abgestufte Rezeptorsequenzähnlichkeit zu konzentrieren, da diese Eigenschaften die entscheidende Brücke zwischen Rezeptorinformation und Zellzustand bilden.
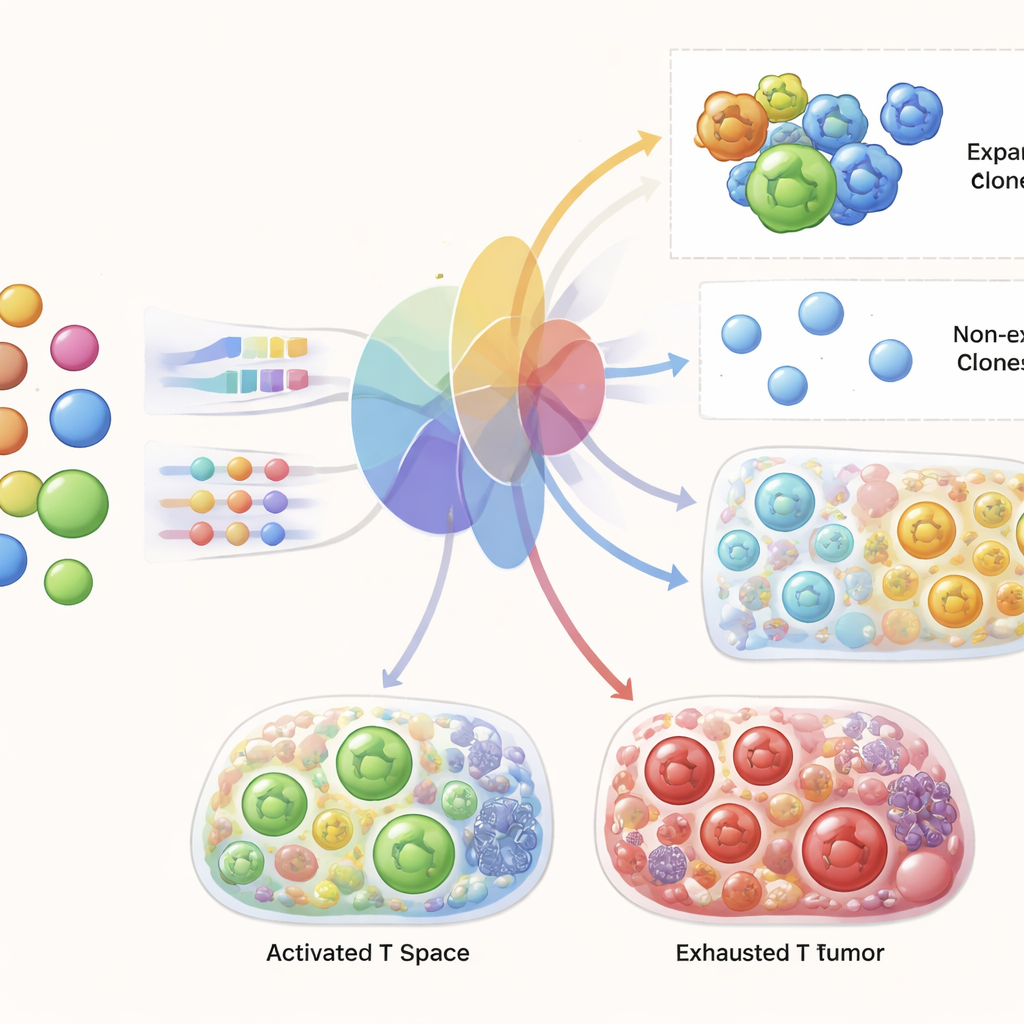
Vorhersage tumorassoziierter T‑Zellen aus dem Blut
Die zentrale Prüfung für TRIM war, ob es allein mithilfe von Blutdaten vor der Behandlung eines Patienten realistische Vorhersagen für dessen T‑Zellen im Tumor und im Blut nach der Therapie treffen kann. Die Autoren trainierten TRIM jeweils auf allen bis auf einen Patienten und fragten dann nach, wie gut das Modell den ausgeklammerten Patienten vorhersagen kann, wobei sie dies über Kohorten hinweg wiederholten. Sie zeigten, dass das Modell die allgemeine Landschaft von T‑Zell‑Genzuständen und Klonvielfalt über Bedingungen hinweg akkurat abbildet. Noch eindrücklicher: Für einzelne Rezeptorklone, die vor der Therapie vorhanden waren, konnte TRIM mit hoher Genauigkeit vorhersagen, welche danach expandieren würden, und übertraf damit klassische Machine‑Learning‑Ansätze. Es reproduzierte zudem bekannte Signaturen erfolgreichen Ansprechens, wie eine Zunahme aktivierter, proliferierender CD8‑T‑Zellen und spezifische Genprogramme, die mit effektivem Tumorzell‑Abtöten verknüpft sind.
Die Gene hinter erfolgreicher Expansion aufdecken
Weil TRIM intern entscheiden muss, welche Zellen wahrscheinlich expandieren, konnten die Autoren das Modell untersuchen, um herauszufinden, auf welche Gene es sich stützt. Die von TRIM hervorgehobenen Gene stimmen mit Kernmerkmalen der T‑Zell‑Aktivierung überein: Zellzyklusregulatoren, die die Proliferation steuern, Moleküle, die am Abtöten von Zielzellen beteiligt sind, Proteine, die Aktivierung und Kommunikation lenken, sowie Stoffwechsel‑ und Migrationsfaktoren, die den T‑Zellen helfen, in Tumoren einzudringen und dort zu funktionieren. Wichtig ist, dass das Modell diese Programme allein mit prätherapeutischen Blutdaten zum Zeitpunkt der Vorhersage identifizierte, was darauf hindeutet, dass frühe molekulare Unterschiede in zirkulierenden T‑Zellen vorhersagen können, wie sie reagieren werden, sobald die Therapie beginnt und sie Tumorsignale begegnen.
Was das für Patientinnen und Patienten bedeutet
Im Kern zeigt diese Arbeit, dass ein sorgfältig gestaltetes Modell aus multimodalen Einzelzelldaten lernen kann, wie die T‑Zellen eines Patienten während einer Krebsimmuntherapie reagieren werden. TRIM nutzt prätherapeutische Blutproben, um abzuleiten, welche Klone expandieren, welche Zustände sie einnehmen und welche Genprogramme dieses Verhalten antreiben — selbst in Geweben, die schwer direkt zu entnehmen sind. Zwar sind weitere Validierung und größere Datensätze vor einem klinischen Einsatz nötig, doch der Ansatz deutet auf eine Zukunft hin, in der Onkologen mithilfe eines einfachen Bluttests in Kombination mit Modellen wie TRIM das Therapieansprechen vorhersagen, den Krankheitsverlauf überwachen und neue Biomarker entdecken könnten, die personalisierte Immuntherapie‑Entscheidungen leiten.
Zitation: He, C., Amodio, M., Ashenberg, O. et al. Multimodal framework for the joint analysis of single-cell RNA and T cell receptor sequencing data predicts T cell response to cancer immunotherapy. Nat Commun 17, 3840 (2026). https://doi.org/10.1038/s41467-026-70505-0
Schlüsselwörter: Krebsimmuntherapie, T‑Zellen, Einzelzellsequenzierung, Machine‑Learning‑Modell, Tumor‑Biomarker