Clear Sky Science · en
Multimodal framework for the joint analysis of single-cell RNA and T cell receptor sequencing data predicts T cell response to cancer immunotherapy
Why predicting immune response matters
Cancer immunotherapies, such as checkpoint inhibitor drugs, work by unleashing a patient’s own T cells to attack tumors. Yet only some patients benefit, and doctors currently have limited tools to forecast who will respond. This study introduces a computational framework called TRIM that learns from detailed single-cell measurements of T cells in the blood and tumors. By combining information about each cell’s gene activity and its unique T cell receptor, TRIM can predict how T cells will expand and change after treatment, offering a potential way to anticipate treatment success from a routine blood draw.
Looking inside individual T cells
T cells patrol the body, searching for signs of danger using their T cell receptors, which recognize specific molecular fragments. When a T cell encounters cancer, it can multiply into a clone of many nearly identical cells and adopt different roles, from active tumor killers to exhausted cells that can no longer fight effectively. Modern single-cell technologies can now read, for each T cell, both its gene expression profile (which genes are turned on or off) and its receptor sequence. The authors applied these tools across thousands of T cells from patients with head and neck, colorectal, and several other cancers, sampling both blood and tumor tissue before and after immunotherapy.
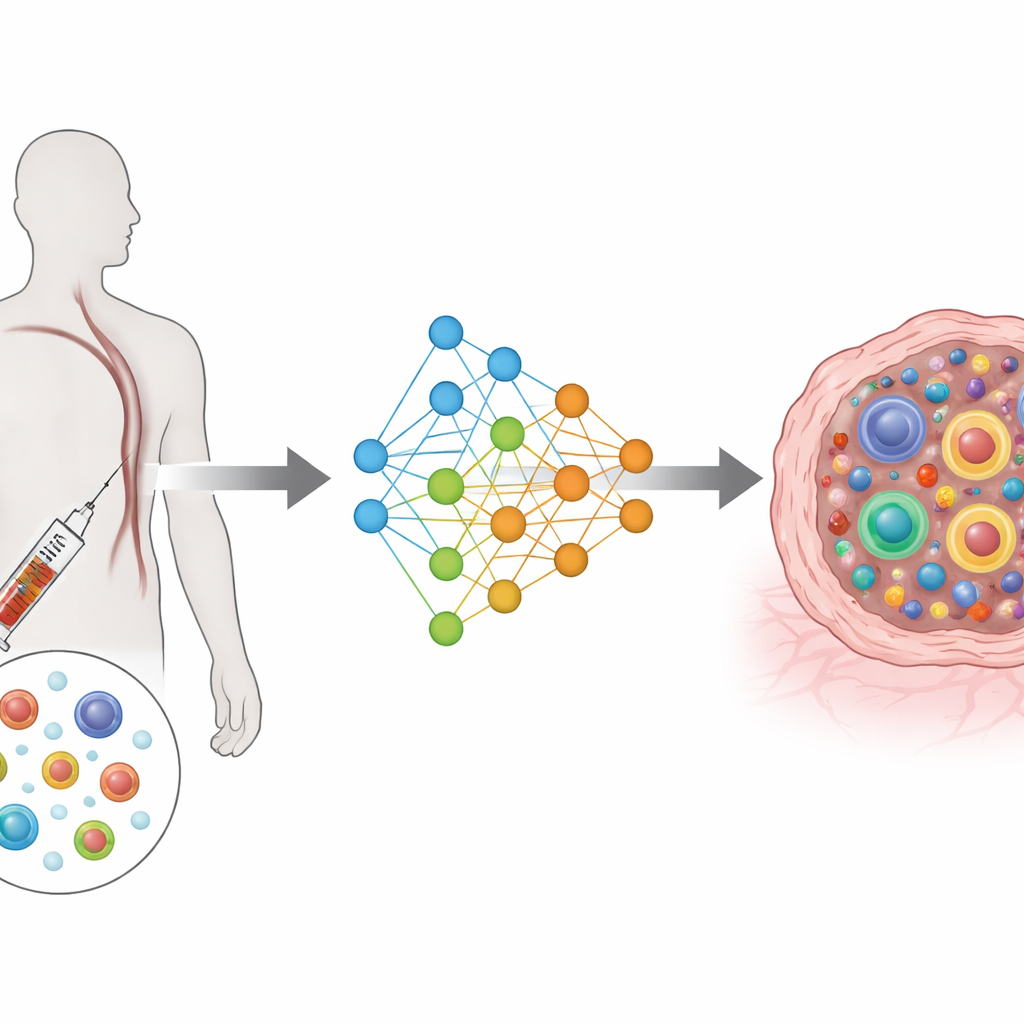
From complex measurements to a shared map
TRIM is built on a type of deep learning architecture known as a conditional variational autoencoder. In simple terms, it compresses the rich information from each cell’s genes and receptor into a shared low-dimensional map, while also taking into account who the patient is, where the cell came from (blood or tumor), and when it was collected (before or after treatment). Separate input modules read the gene activity and receptor features, and separate output modules try to reconstruct them. A special loss function nudges cells that belong to the same clone to sit close together in this map, while keeping different clones apart. This design focuses the model on what matters most biologically: how large each clone is, and how its cells are distributed across functional states.
What links receptors, gene activity, and clones
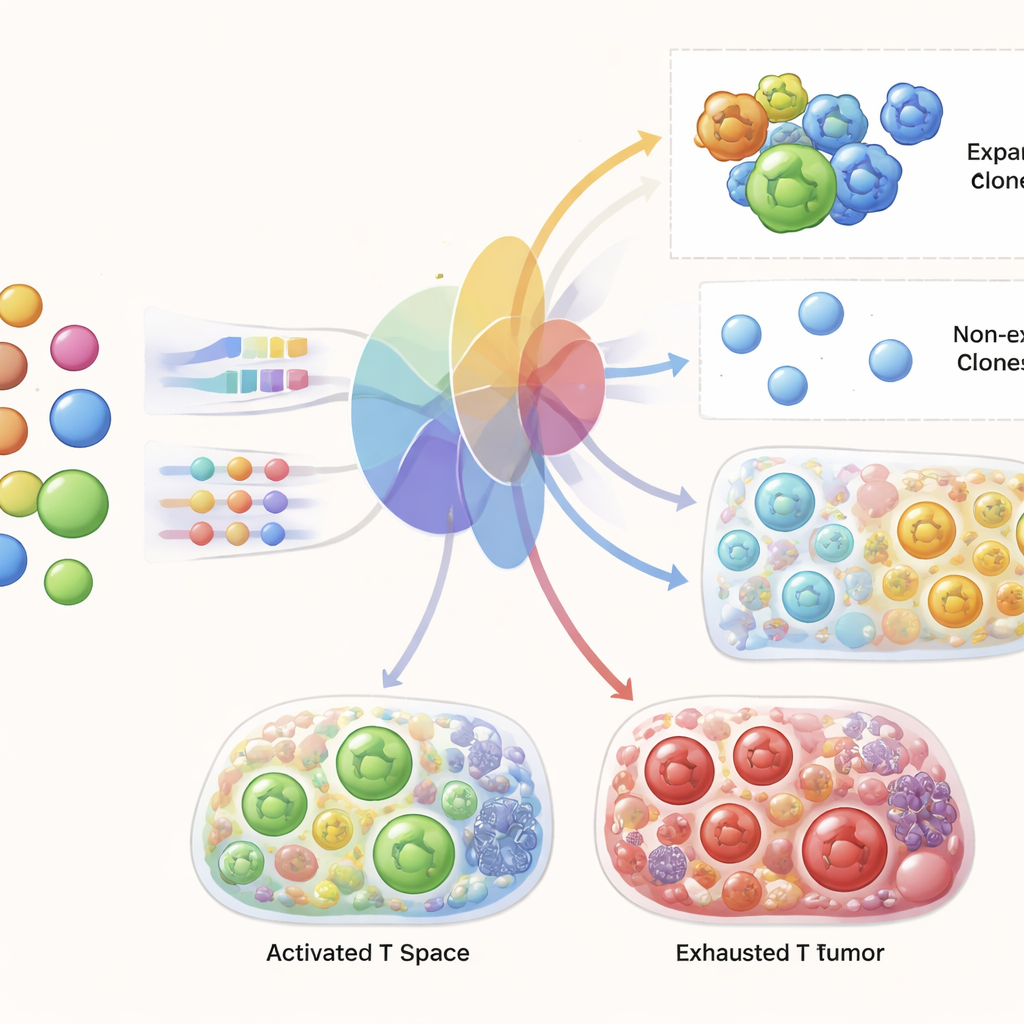
By systematically comparing receptor similarity and gene expression across multiple datasets, the researchers found that cells sharing exactly the same receptor are indeed more similar to each other in gene activity than random T cells, particularly for large, expanded clones. However, beyond exact matches, small changes in the receptor sequence do not reliably track with how similar two cells are in their gene expression. In other words, cells with nearly identical receptors can look as different as cells with completely unrelated receptors. This led the team to concentrate on clone identity and clone size, rather than fine-grained receptor sequence similarity, as the crucial bridge between receptor information and cell state.
Predicting tumor-bound T cells from blood
The central test for TRIM was whether it could take only pre-treatment blood data from a patient and generate realistic predictions for that patient’s T cells in the tumor and in blood after therapy. The authors trained TRIM on all but one patient and then asked it to predict the held-out patient, repeating this across cohorts. They showed that the model accurately captures the overall landscape of T cell gene states and clone diversity across conditions. More strikingly, for individual receptor clones present before therapy, TRIM could predict which ones would expand after treatment with high accuracy, clearly outperforming standard machine-learning approaches. It also reproduced known signatures of successful response, such as an increase in activated, proliferating CD8 T cells and specific gene programs linked to effective tumor killing.
Uncovering the genes behind successful expansion
Because TRIM must internally decide which cells are likely to expand, the authors could probe the model to see which genes it relies on. The genes flagged by TRIM align with core features of T cell activation: cell-cycle regulators that control proliferation, molecules involved in killing target cells, proteins that govern activation and communication, and metabolic and migration-related factors that help T cells move into and function within tumors. Importantly, the model identified these programs using only pre-treatment blood data at prediction time, suggesting that early molecular differences in circulating T cells foreshadow how they will respond once therapy begins and they encounter tumor signals.
What this means for patients
In essence, this work shows that a carefully designed model can learn from multimodal single-cell data to forecast how a patient’s T cells will behave during cancer immunotherapy. TRIM uses pre-treatment blood samples to infer which clones will expand, what states they will enter, and which gene programs will drive this behavior, even in tissues that are difficult to sample directly. While further validation and larger datasets are needed before clinical use, the approach points toward a future where oncologists might use a simple blood test, paired with models like TRIM, to predict treatment response, monitor disease progression, and discover new biomarkers that guide personalized immunotherapy decisions.
Citation: He, C., Amodio, M., Ashenberg, O. et al. Multimodal framework for the joint analysis of single-cell RNA and T cell receptor sequencing data predicts T cell response to cancer immunotherapy. Nat Commun 17, 3840 (2026). https://doi.org/10.1038/s41467-026-70505-0
Keywords: cancer immunotherapy, T cells, single-cell sequencing, machine learning model, tumor biomarkers