Clear Sky Science · it
Quadro multimodale per l’analisi congiunta di dati single-cell RNA e di sequenziamento del recettore delle cellule T predice la risposta delle cellule T all’immunoterapia oncologica
Perché prevedere la risposta immunitaria è importante
Le immunoterapie contro il cancro, come gli inibitori dei checkpoint, agiscono liberando le cellule T del paziente per attaccare i tumori. Tuttavia solo alcuni pazienti ne traggono beneficio e i medici hanno attualmente strumenti limitati per prevedere chi risponderà. Questo studio presenta un quadro computazionale chiamato TRIM che apprende da misure single-cell dettagliate delle cellule T nel sangue e nei tumori. Combinando informazioni sull’attività genica di ciascuna cellula e sul suo recettore T unico, TRIM può prevedere come le cellule T si espanderanno e si trasformeranno dopo il trattamento, offrendo una possibile via per anticipare il successo della terapia a partire da un semplice prelievo di sangue.
Uno sguardo all’interno delle singole cellule T
Le cellule T pattugliano il corpo, cercando segnali di pericolo tramite i loro recettori, che riconoscono specifici frammenti molecolari. Quando una cellula T incontra il cancro, può moltiplicarsi in un clone di molte cellule quasi identiche e assumere ruoli diversi, da killer tumorali attivi a cellule esauste che non riescono più a combattere efficacemente. Le tecnologie single-cell moderne possono ora leggere, per ogni cellula T, sia il profilo di espressione genica (quali geni sono attivi o spenti) sia la sequenza del recettore. Gli autori hanno applicato questi strumenti a migliaia di cellule T provenienti da pazienti con tumori della testa e del collo, del colon-retto e altri tipi, campionando sia il sangue sia il tessuto tumorale prima e dopo l’immunoterapia.
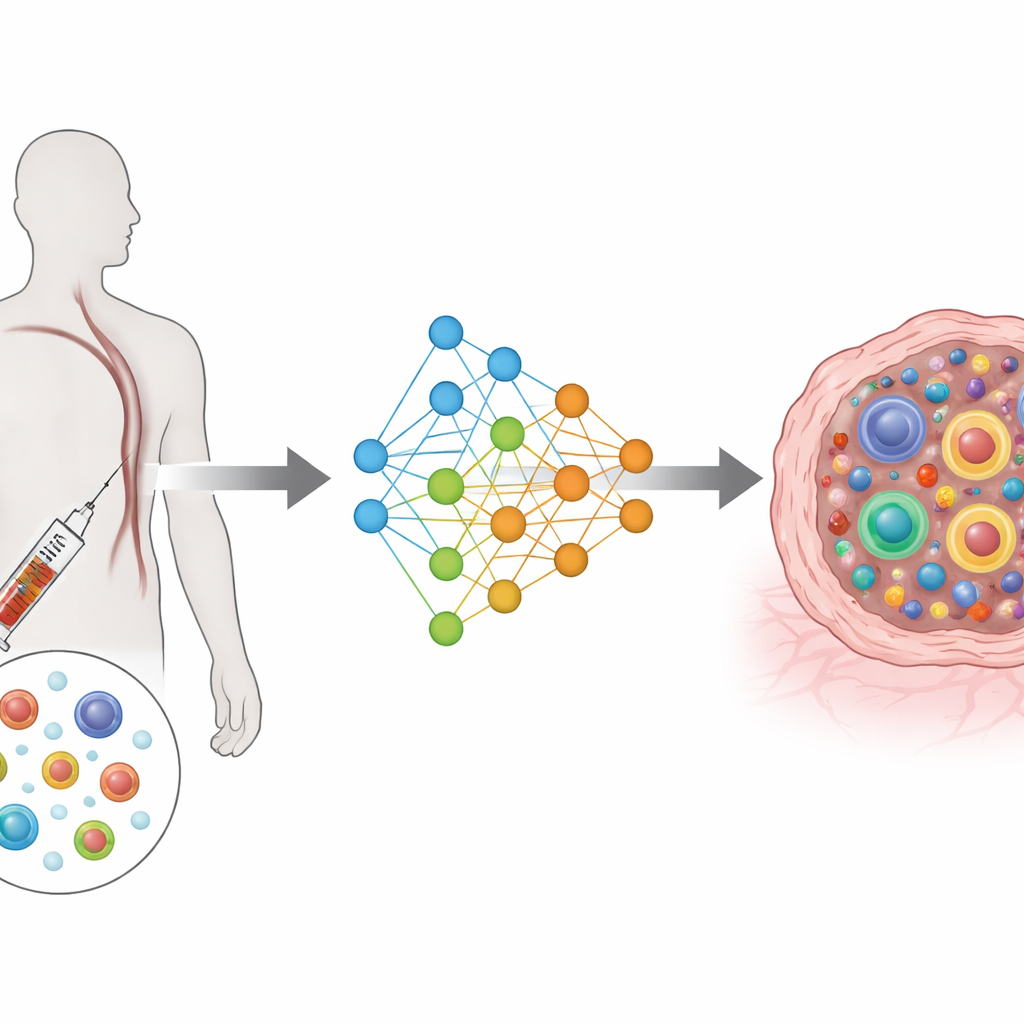
Da misure complesse a una mappa condivisa
TRIM si basa su un’architettura di deep learning nota come autoencoder variazionale condizionato. In termini semplici, comprime le informazioni ricche provenienti dai geni e dai recettori di ciascuna cellula in una mappa condivisa a bassa dimensionalità, tenendo conto anche del paziente, della provenienza della cellula (sangue o tumore) e del momento del prelievo (prima o dopo il trattamento). Moduli di input separati leggono l’attività genica e le caratteristiche del recettore, mentre moduli di output separati cercano di ricostruirle. Una funzione di perdita speciale spinge le cellule appartenenti allo stesso clone a disporsi vicine in questa mappa, mantenendo invece separati i cloni diversi. Questo progetto concentra il modello su ciò che biologicamente conta di più: quanto è grande ciascun clone e come le sue cellule sono distribuite nei diversi stati funzionali.
Cosa collega recettori, attività genica e cloni
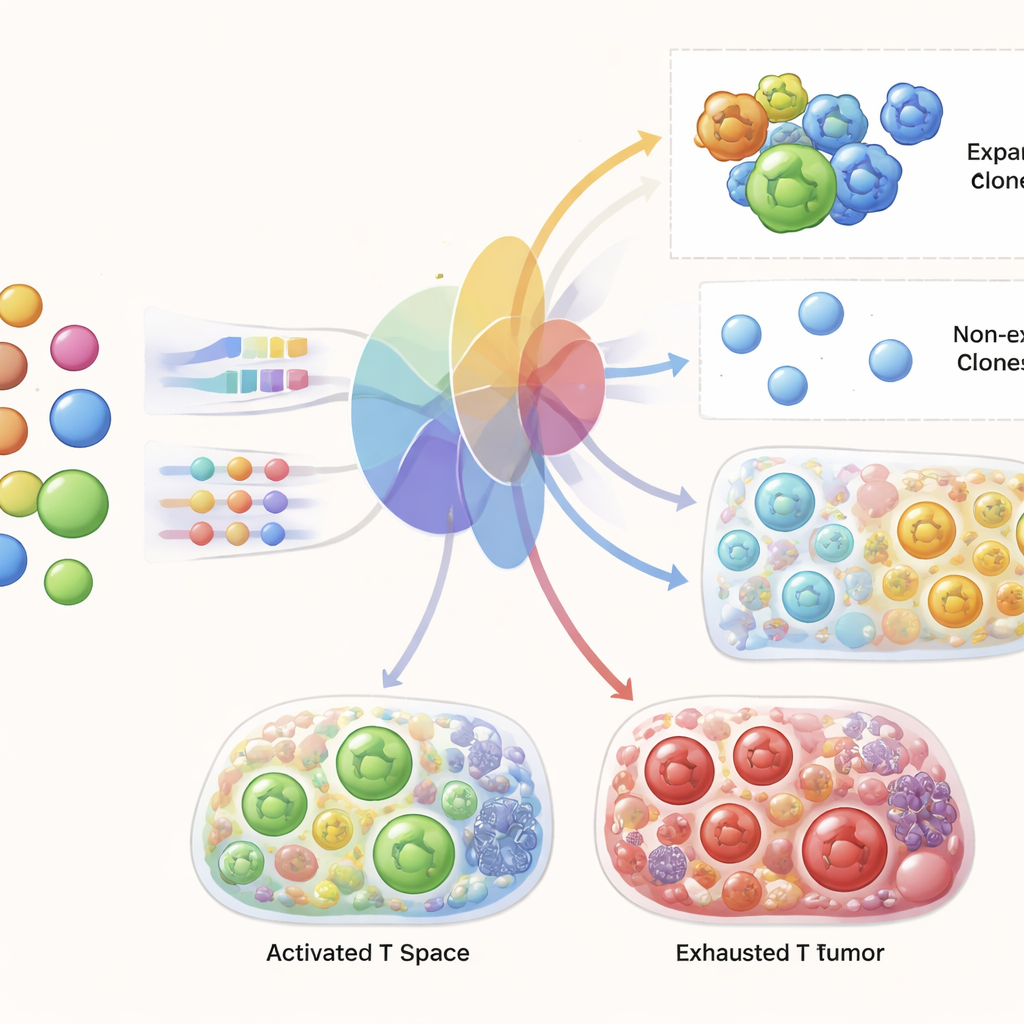
Confrontando sistematicamente la somiglianza dei recettori e l’espressione genica attraverso più dataset, i ricercatori hanno constatato che le cellule che condividono esattamente lo stesso recettore sono effettivamente più simili tra loro nell’attività genica rispetto a cellule T casuali, in particolare per i cloni grandi ed espansi. Tuttavia, al di là delle corrispondenze esatte, piccole variazioni nella sequenza del recettore non corrispondono in modo affidabile alla somiglianza nell’espressione genica tra due cellule. In altre parole, cellule con recettori quasi identici possono apparire tanto diverse quanto cellule con recettori completamente non correlati. Questo ha portato il team a concentrarsi sull’identità del clone e sulla dimensione del clone, piuttosto che sulla somiglianza fine della sequenza del recettore, come ponte cruciale tra informazione sul recettore e stato cellulare.
Prevedere le cellule T legate al tumore a partire dal sangue
Il test centrale per TRIM è stato verificare se poteva prendere solo i dati del sangue pre-trattamento di un paziente e generare previsioni realistiche per le cellule T di quel paziente nel tumore e nel sangue dopo la terapia. Gli autori hanno addestrato TRIM escludendo un paziente alla volta e poi hanno chiesto al modello di prevedere il paziente tenuto fuori, ripetendo questa procedura attraverso le coorti. Hanno mostrato che il modello cattura accuratamente il panorama complessivo degli stati genici delle cellule T e della diversità dei cloni nelle diverse condizioni. Ancora più impressionante, per singoli cloni di recettori presenti prima della terapia, TRIM è stato in grado di prevedere quali si sarebbero espansi dopo il trattamento con alta accuratezza, superando nettamente gli approcci standard di machine learning. Ha inoltre riprodotto firme note di risposta efficace, come l’aumento di cellule CD8 attivate e in proliferazione e programmi genici specifici legati all’uccisione tumorale efficace.
Scoprire i geni dietro l’espansione riuscita
Poiché TRIM deve internamente decidere quali cellule sono probabili candidate all’espansione, gli autori hanno potuto sondare il modello per vedere su quali geni si basa. I geni evidenziati da TRIM corrispondono alle caratteristiche fondamentali dell’attivazione delle cellule T: regolatori del ciclo cellulare che controllano la proliferazione, molecole coinvolte nell’uccisione delle cellule bersaglio, proteine che governano attivazione e comunicazione, e fattori metabolici e di migrazione che aiutano le cellule T a infiltrarsi e funzionare all’interno dei tumori. Importante è che il modello ha identificato questi programmi usando solo i dati del sangue pre-trattamento al momento della predizione, suggerendo che differenze molecolari precoci nelle cellule T circolanti preannunciano come risponderanno una volta che la terapia comincia e incontrano segnali tumorali.
Cosa significa per i pazienti
In sostanza, questo lavoro mostra che un modello accuratamente progettato può apprendere da dati multimodali single-cell per prevedere come si comporteranno le cellule T di un paziente durante l’immunoterapia oncologica. TRIM usa campioni di sangue pre-trattamento per inferire quali cloni si espanderanno, in quali stati entreranno e quali programmi genici guideranno questo comportamento, anche in tessuti difficili da campionare direttamente. Sebbene siano necessarie ulteriori validazioni e dataset più ampi prima dell’uso clinico, l’approccio indica un futuro in cui gli oncologi potrebbero usare un semplice esame del sangue, affiancato a modelli come TRIM, per prevedere la risposta alla terapia, monitorare la progressione della malattia e scoprire nuovi biomarcatori che guidino decisioni di immunoterapia personalizzata.
Citazione: He, C., Amodio, M., Ashenberg, O. et al. Multimodal framework for the joint analysis of single-cell RNA and T cell receptor sequencing data predicts T cell response to cancer immunotherapy. Nat Commun 17, 3840 (2026). https://doi.org/10.1038/s41467-026-70505-0
Parole chiave: immunoterapia oncologica, cellule T, sequenziamento single-cell, modello di machine learning, biomarcatori tumorali