Clear Sky Science · es
Marco multimodal para el análisis conjunto de datos de secuenciación de ARN de célula única y de receptores de células T predice la respuesta de las células T a la inmunoterapia contra el cáncer
Por qué importa predecir la respuesta inmunitaria
Las inmunoterapias contra el cáncer, como los inhibidores de puntos de control, actúan liberando las propias células T del paciente para que ataquen los tumores. Sin embargo, solo algunos pacientes se benefician y los médicos disponen actualmente de herramientas limitadas para prever quién responderá. Este estudio presenta un marco computacional denominado TRIM que aprende a partir de mediciones de célula única detalladas de células T en sangre y tumores. Al combinar información sobre la actividad génica de cada célula y su receptor único de célula T, TRIM puede predecir cómo se expandirán y cambiarán las células T tras el tratamiento, ofreciendo una vía potencial para anticipar el éxito terapéutico a partir de una extracción de sangre de rutina.
Mirando dentro de células T individuales
Las células T patrullan el organismo buscando señales de peligro mediante sus receptores de células T, que reconocen fragmentos moleculares específicos. Cuando una célula T se encuentra con cáncer, puede multiplicarse formando un clon de muchas células casi idénticas y adoptar distintos roles, desde asesinos tumorales activos hasta células agotadas que ya no pueden combatir eficazmente. Las tecnologías modernas de célula única pueden ahora leer, para cada célula T, tanto su perfil de expresión génica (qué genes están activados o desactivados) como la secuencia de su receptor. Los autores aplicaron estas herramientas a miles de células T procedentes de pacientes con cánceres de cabeza y cuello, colorrectal y otros tipos, muestreando tanto sangre como tejido tumoral antes y después de la inmunoterapia.
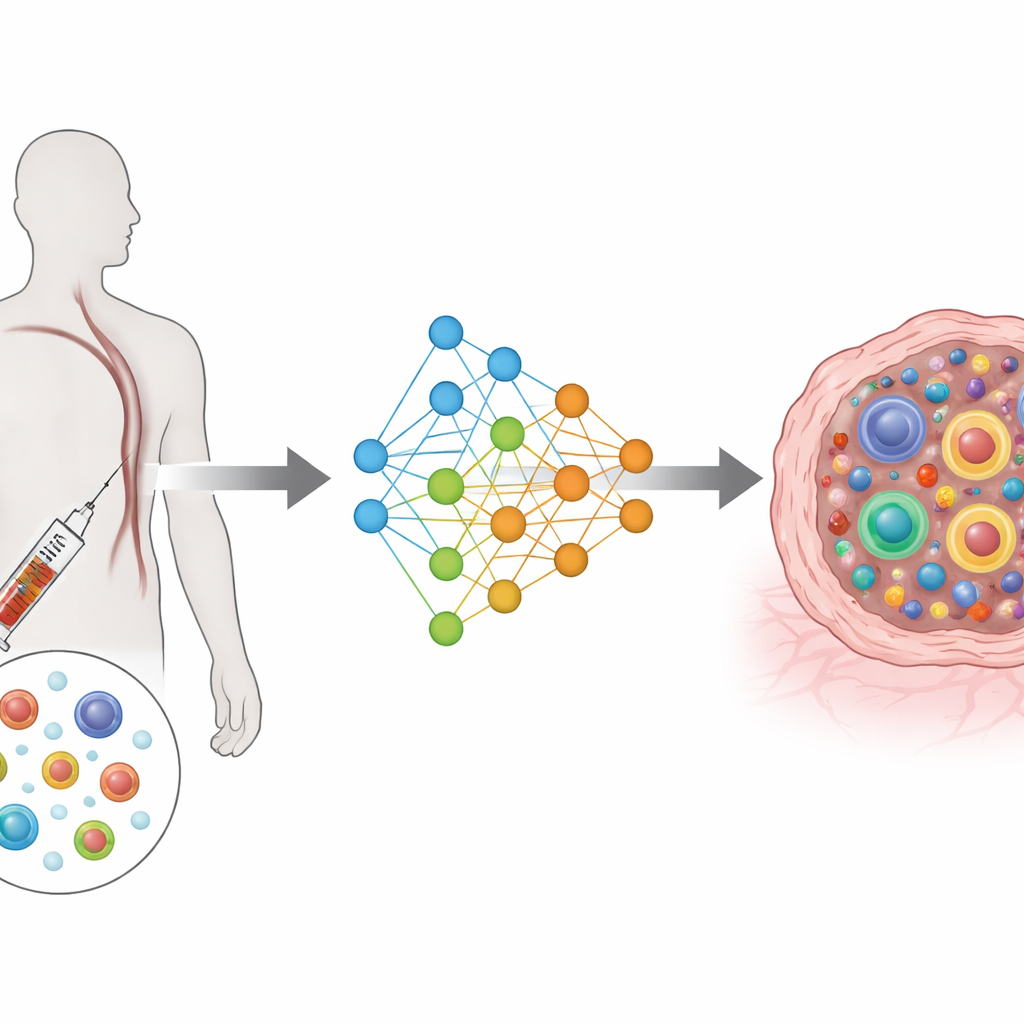
De mediciones complejas a un mapa compartido
TRIM se basa en un tipo de arquitectura de aprendizaje profundo conocida como autoencoder variacional condicional. En términos sencillos, comprime la información rica de los genes y del receptor de cada célula en un mapa compartido de baja dimensión, teniendo en cuenta además quién es el paciente, de dónde proviene la célula (sangre o tumor) y cuándo se recogió (antes o después del tratamiento). Módulos de entrada separados leen la actividad génica y las características del receptor, y módulos de salida separados intentan reconstruirlas. Una función de pérdida especial empuja a que las células pertenecientes al mismo clon queden próximas en ese mapa, manteniendo a la vez separados a los clones diferentes. Este diseño centra el modelo en lo que biológicamente importa: el tamaño de cada clon y cómo se distribuyen sus células entre estados funcionales.
Qué vincula receptores, actividad génica y clones
Al comparar sistemáticamente la similitud de receptores y la expresión génica en múltiples conjuntos de datos, los investigadores encontraron que las células que comparten exactamente el mismo receptor son, efectivamente, más similares entre sí en su actividad génica que células T al azar, en particular en clones grandes y expandidos. Sin embargo, más allá de las coincidencias exactas, pequeños cambios en la secuencia del receptor no se correlacionan de forma fiable con la similitud en la expresión génica entre dos células. En otras palabras, células con receptores casi idénticos pueden parecer tan diferentes como células con receptores completamente no relacionados. Esto llevó al equipo a centrarse en la identidad del clon y en el tamaño del clon, más que en la similitud de secuencia de receptor a nivel fino, como el puente crucial entre la información del receptor y el estado celular.
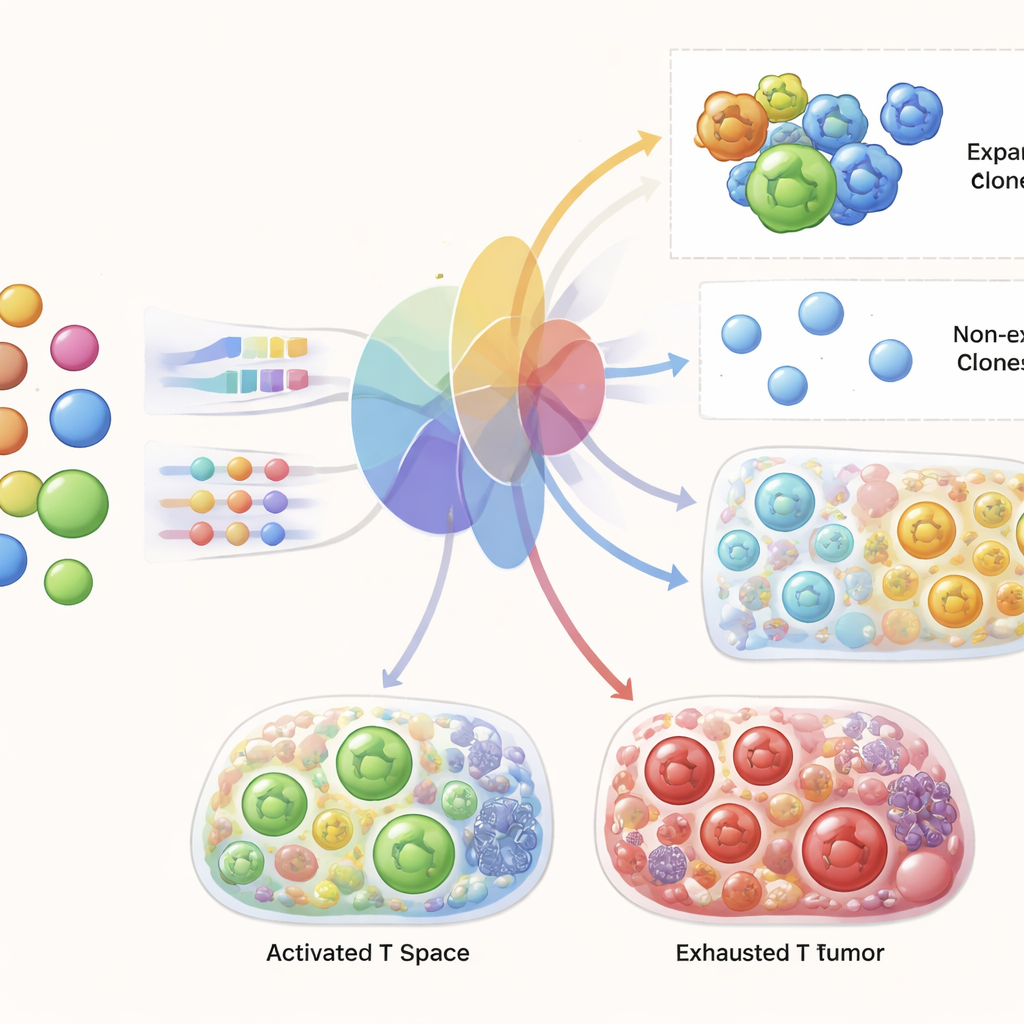
Predecir células T ligadas al tumor a partir de sangre
La prueba central para TRIM fue si podía tomar solo datos de sangre previos al tratamiento de un paciente y generar predicciones realistas para las células T de ese paciente en el tumor y en la sangre después de la terapia. Los autores entrenaron TRIM con todos los pacientes salvo uno y luego le pidieron predecir al paciente excluido, repitiendo este procedimiento entre las cohortes. Mostraron que el modelo captura con precisión el paisaje general de estados génicos de las células T y la diversidad clonal entre condiciones. Más llamativo aún, para clones de receptores individuales presentes antes de la terapia, TRIM pudo predecir cuáles se expandirían después del tratamiento con alta precisión, superando claramente a enfoques estándar de aprendizaje automático. También reprodujo firmas conocidas de respuesta exitosa, como un aumento en células CD8 activadas y proliferantes y programas génicos específicos vinculados a la eliminación eficaz del tumor.
Descubriendo los genes detrás de la expansión exitosa
Como TRIM debe decidir internamente qué células probablemente se expandirán, los autores pudieron sondear el modelo para ver en qué genes se apoya. Los genes señalados por TRIM coinciden con características centrales de la activación de células T: reguladores del ciclo celular que controlan la proliferación, moléculas implicadas en la eliminación de células diana, proteínas que gobiernan la activación y la comunicación, y factores metabólicos y relacionados con la migración que ayudan a las células T a desplazarse y funcionar dentro de los tumores. Es importante que el modelo identificara estos programas usando únicamente datos de sangre previos al tratamiento en el momento de la predicción, lo que sugiere que diferencias moleculares tempranas en las células T circulantes anticipan cómo responderán una vez que comience la terapia y entren en contacto con señales tumorales.
Qué significa esto para los pacientes
En esencia, este trabajo demuestra que un modelo diseñado con cuidado puede aprender de datos multimodales de célula única para prever cómo se comportarán las células T de un paciente durante la inmunoterapia contra el cáncer. TRIM utiliza muestras de sangre previas al tratamiento para inferir qué clones se expandirán, qué estados adoptarán y qué programas génicos impulsarán ese comportamiento, incluso en tejidos difíciles de muestrear directamente. Aunque son necesarias más validaciones y conjuntos de datos más grandes antes de un uso clínico, el enfoque apunta hacia un futuro en el que los oncólogos podrían emplear una simple prueba de sangre, junto con modelos como TRIM, para predecir la respuesta al tratamiento, monitorizar la progresión de la enfermedad y descubrir nuevos biomarcadores que guíen decisiones de inmunoterapia personalizadas.
Cita: He, C., Amodio, M., Ashenberg, O. et al. Multimodal framework for the joint analysis of single-cell RNA and T cell receptor sequencing data predicts T cell response to cancer immunotherapy. Nat Commun 17, 3840 (2026). https://doi.org/10.1038/s41467-026-70505-0
Palabras clave: inmunoterapia contra el cáncer, células T, secuenciación de célula única, modelo de aprendizaje automático, biomarcadores tumorales