Clear Sky Science · pt
Percepções moleculares sobre a síntese fúngica de inositol fosforilceramida e sua inibição pelo antifúngico aureobasidina A
Por que essa história fúngica importa
Infecções fúngicas invasivas matam discretamente milhões de pessoas a cada ano, especialmente as com sistemas imunológicos enfraquecidos. Ainda assim, os médicos dispõem de apenas um punhado de antifúngicos, e muitos fungos vêm desenvolvendo resistência. Este estudo concentra-se em um processo fúngico que não existe em células humanas, tornando‑o um alvo ideal para novos medicamentos. Ao desvendar como uma enzima fúngica chave funciona — e como um fármaco potente a bloqueia — os pesquisadores estabelecem bases para terapias antifúngicas mais seguras e eficazes.
Um ponto fraco único na armadura fúngica
Células fúngicas dependem de uma classe especial de lipídios, chamados esfingolipídios, para construir e organizar suas membranas. Um desses, a inositol fosforilceramida (IPC), é encontrado em fungos, mas ausente em mamíferos. A IPC é produzida por um complexo enzimático de duas partes conhecido como sintetase de IPC. Um parceiro, Aur1, realiza a química; o outro, Kei1, ajuda a posicionar e estabilizar o complexo na membrana. Porque fungos não podem viver sem a sintetase de IPC, enquanto os humanos não a possuem, essa enzima há muito é vista como um alvo atraente para fármacos.
Um antifúngico potente e um alvo misterioso
O composto aureobasidina A, usado principalmente em pesquisa até agora, é um dos antifúngicos mais potentes conhecidos. Ele pode matar patógenos fúngicos importantes em doses minúsculas e mostra baixa toxicidade para células de mamíferos. Trabalhos genéticos já haviam mostrado que alterações na proteína Aur1 podem tornar fungos resistentes ao fármaco, confirmando que a sintetase de IPC é seu alvo. Mas sem uma visão em alta resolução da enzima, os cientistas não conseguiam ver exatamente como ela reconhece seus substratos lipídicos naturais nem como a aureobasidina A se encaixa para interromper a reação.
Criando imagens da enzima em ação
Para resolver esse problema, os pesquisadores utilizaram criomicroscopia eletrônica, uma técnica que imagina moléculas congeladas instantaneamente com detalhe quase atômico. A sintetase de IPC é pequena e em grande parte enterrada na membrana, o que dificulta sua visualização. A equipe superou isso anexando uma pequena etiqueta proteica e um nanocorpo combinante como “alça” que melhorou o alinhamento das imagens sem perturbar a atividade da enzima ou sua sensibilidade ao fármaco. Em seguida, determinaram estruturas da sintetase de IPC de levedura em dois estados chave: ligada ao seu substrato lipídico ceramida, e ligada à aureobasidina A.
A cavidade de trabalho da enzima e seu parceiro de controle
As estruturas revelam Aur1 e Kei1 formando um par compacto que atravessa a membrana. Aur1 contém uma câmara de reação em forma de sulco que se abre lateralmente para os lipídios circundantes. A ceramida se aloja nesse sulco, com uma cauda enterrada profundamente ao longo de uma linha de aminoácidos apolares e sua cabeça reativa apontando para três resíduos cruciais — duas histidinas e um aspartato — que se situam na base da câmara. Mutar qualquer membro dessa “tríade H–H–D” aboliu completamente a atividade enzimática, mostrando que juntos realizam a transferência em dois passos de um grupo portador de fósforo de um lipídio para outro.
Kei1, antes considerado um ajudante passivo, mostra‑se essencial. Ele forma contatos extensos com uma alça de Aur1 e envolve um bolso compartilhado que liga firmemente uma molécula de fosfolipídio. Quando resíduos chave que prendem esse lipídio são mutados, o complexo Aur1–Kei1 se desfaz e a atividade é perdida. Isso sugere que lipídios nativos da membrana agem como cunhas estruturais que estabilizam o complexo enzimático, apontando para maneiras adicionais pelas quais drogas podem perturbar sua função.
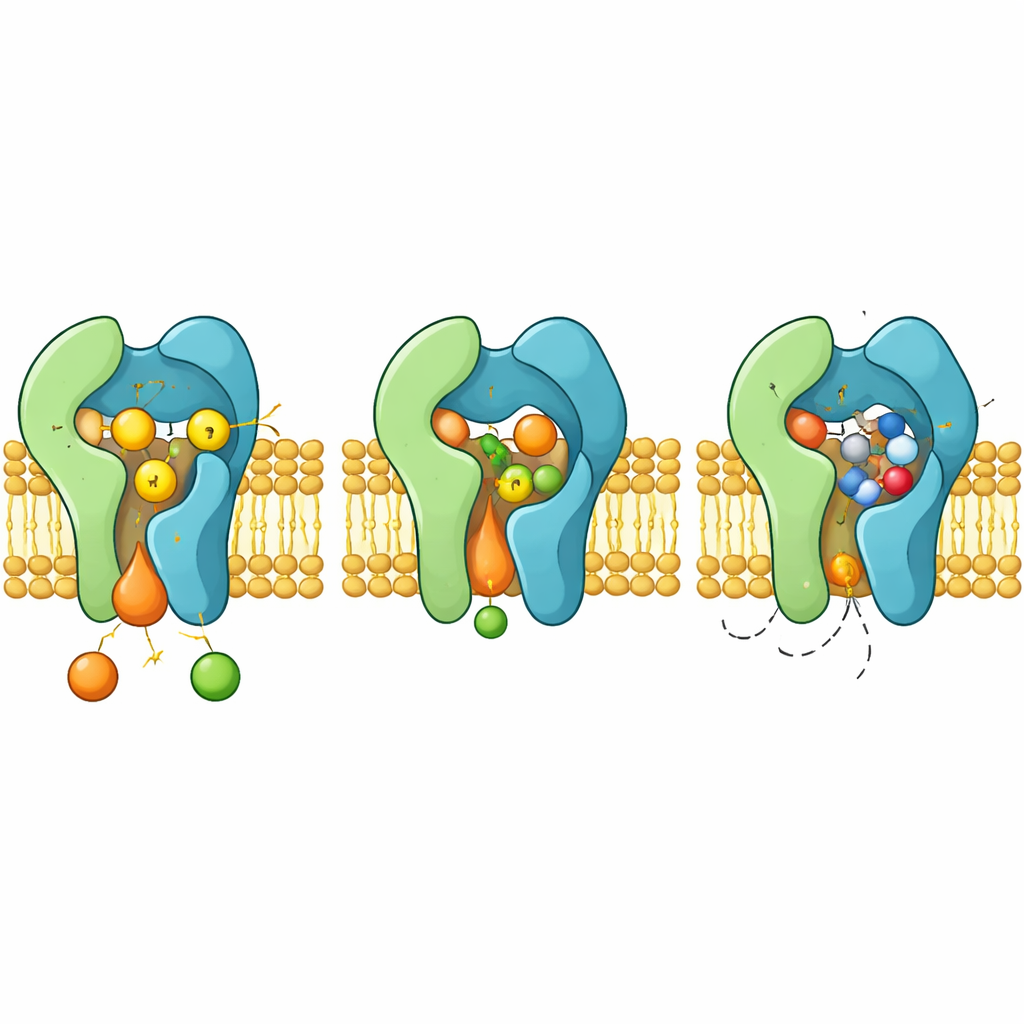
Como o fármaco bloqueia e como surge a resistência
Quando a aureobasidina A se liga, ela ocupa a mesma câmara de reação que normalmente acomoda a ceramida. A molécula cíclica se encaixa entre alças e hélices flexíveis, empurrando de lado cadeias laterais aromáticas que se reorientam para criar um bolso justo. Nessa configuração, o fármaco oclui a tríade H–H–D e exclui estéricamente a ceramida, agindo como um inibidor competitivo clássico. As estruturas também explicam como mutações específicas em Aur1, como alterações nos resíduos H157 ou F158 próximos ao bolso, enfraquecem a ligação do fármaco ao mesmo tempo em que preservam a catálise, dando origem a fungos resistentes.
O que isso significa para futuros antifúngicos
Em conjunto, esses achados fornecem um mapa detalhado de uma enzima da qual fungos não podem prescindir e que os humanos não possuem. Ao mostrar exatamente como a sintetase de IPC se monta, como reconhece seus substratos lipídicos e como a aureobasidina A se enfia em sua câmara de reação, o estudo oferece um roteiro para projetar novos fármacos que desabilitem seletivamente as membranas fúngicas. Compostos guiados por estruturas assim poderiam expandir o arsenal antifúngico limitado e ajudar a superar o problema crescente de infecções fúngicas resistentes a medicamentos.
Citação: Chen, J., Ke, Y., Zhang, M. et al. Molecular insights into fungal inositol phosphorylceramide synthesis and its inhibition by antifungal aureobasidin A. Nat Commun 17, 3951 (2026). https://doi.org/10.1038/s41467-026-69777-3
Palavras-chave: medicamentos antifúngicos, membrana celular fúngica, esfingolipídios, resistência a medicamentos, criomicroscopia eletrônica