Clear Sky Science · en
Molecular insights into fungal inositol phosphorylceramide synthesis and its inhibition by antifungal aureobasidin A
Why this fungus story matters
Invasive fungal infections quietly kill millions of people each year, especially those with weakened immune systems. Yet doctors have only a handful of antifungal drugs, and many fungi are evolving resistance. This study focuses on a fungal process that does not exist in human cells, making it an ideal bullseye for new medicines. By uncovering how a key fungal enzyme works—and how a powerful drug jams it—the researchers lay groundwork for safer, more effective antifungal therapies.
A unique weak spot in fungal armor
Fungal cells rely on a special class of fats, called sphingolipids, to build and organize their membranes. One of these, inositol phosphorylceramide (IPC), is found in fungi but absent in mammals. IPC is made by a two-part enzyme complex known as IPC synthase. One partner, Aur1, does the chemistry; the other, Kei1, helps position and stabilize the complex in the membrane. Because fungi cannot live without IPC synthase, while humans do not have it at all, this enzyme has long been viewed as an attractive drug target.
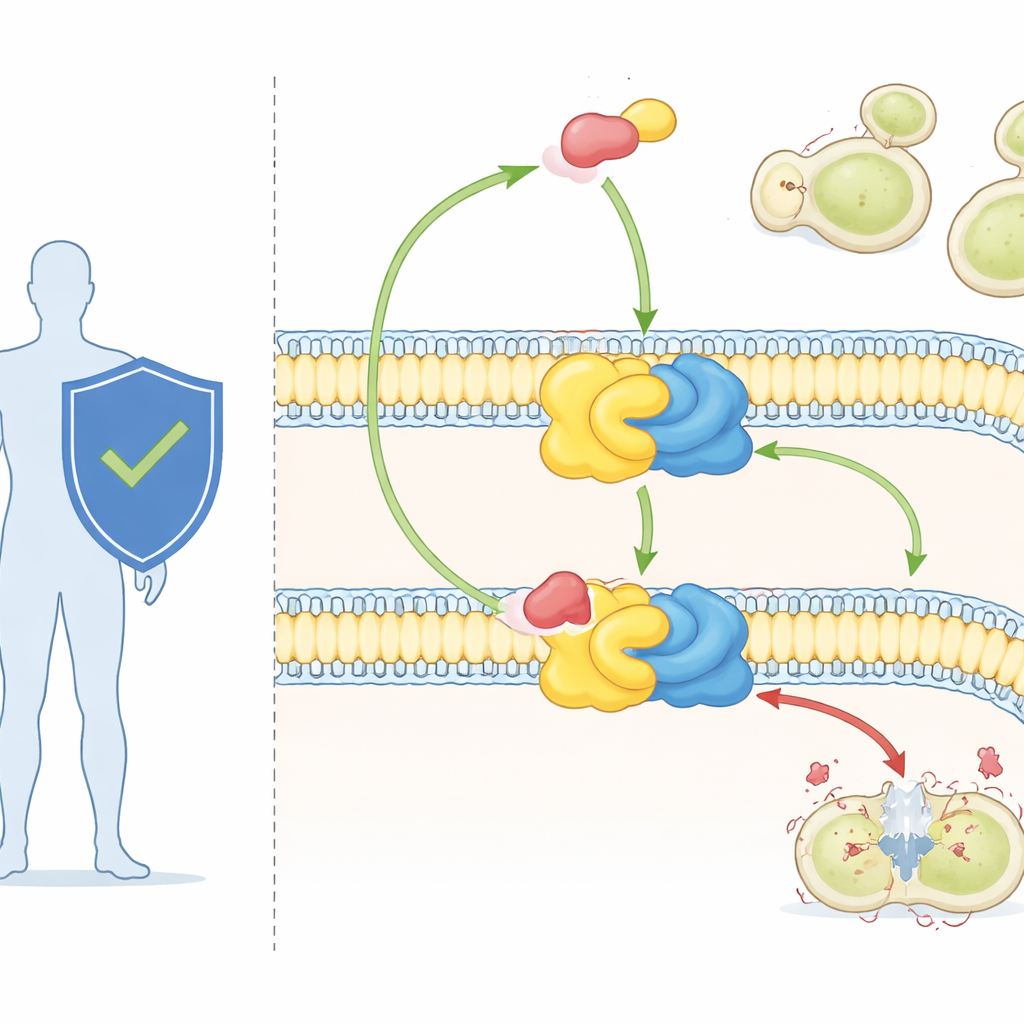
A powerful antifungal and a mysterious target
The drug aureobasidin A, used mainly in research so far, is one of the most potent known antifungals. It can kill major fungal pathogens at tiny doses and shows little toxicity to mammalian cells. Genetic work had already shown that changes in the Aur1 protein can make fungi resistant to the drug, confirming that IPC synthase is its target. But without a high-resolution view of the enzyme, scientists could not see exactly how it recognizes its natural lipid substrates or how aureobasidin A wedges itself in to shut the reaction down.
Freezing the enzyme in action
To solve this problem, the researchers used cryo–electron microscopy, a technique that images flash-frozen molecules at near‑atomic detail. IPC synthase is small and mostly buried in the membrane, which makes it hard to visualize. The team overcame this by attaching a tiny protein tag and a matching nanobody “handle” that improved image alignment without disrupting the enzyme’s activity or its sensitivity to the drug. They then determined structures of the yeast IPC synthase in two key states: bound to its lipid substrate ceramide, and bound to aureobasidin A.
The enzyme’s working cavity and control partner
The structures reveal Aur1 and Kei1 forming a tight pair spanning the membrane. Aur1 contains a groove-like reaction chamber that opens sideways into the surrounding lipids. Ceramide nestles into this groove, with one tail buried deep along a line of oily amino acids and its reactive head pointing toward three crucial residues—two histidines and one aspartate—that sit at the base of the chamber. Mutating any member of this “H–H–D triad” completely abolishes enzyme activity, showing that together they carry out the two-step transfer of a phosphate-bearing group from one lipid to another.
Kei1, once thought to be a passive helper, turns out to be essential. It forms extensive contacts with a loop from Aur1 and embraces a shared pocket that tightly binds a phospholipid molecule. When key residues that hold this lipid in place are mutated, the Aur1–Kei1 assembly falls apart and activity is lost. This suggests that native membrane lipids act as structural wedges that stabilize the enzyme complex, pointing to additional ways drugs might disrupt its function.
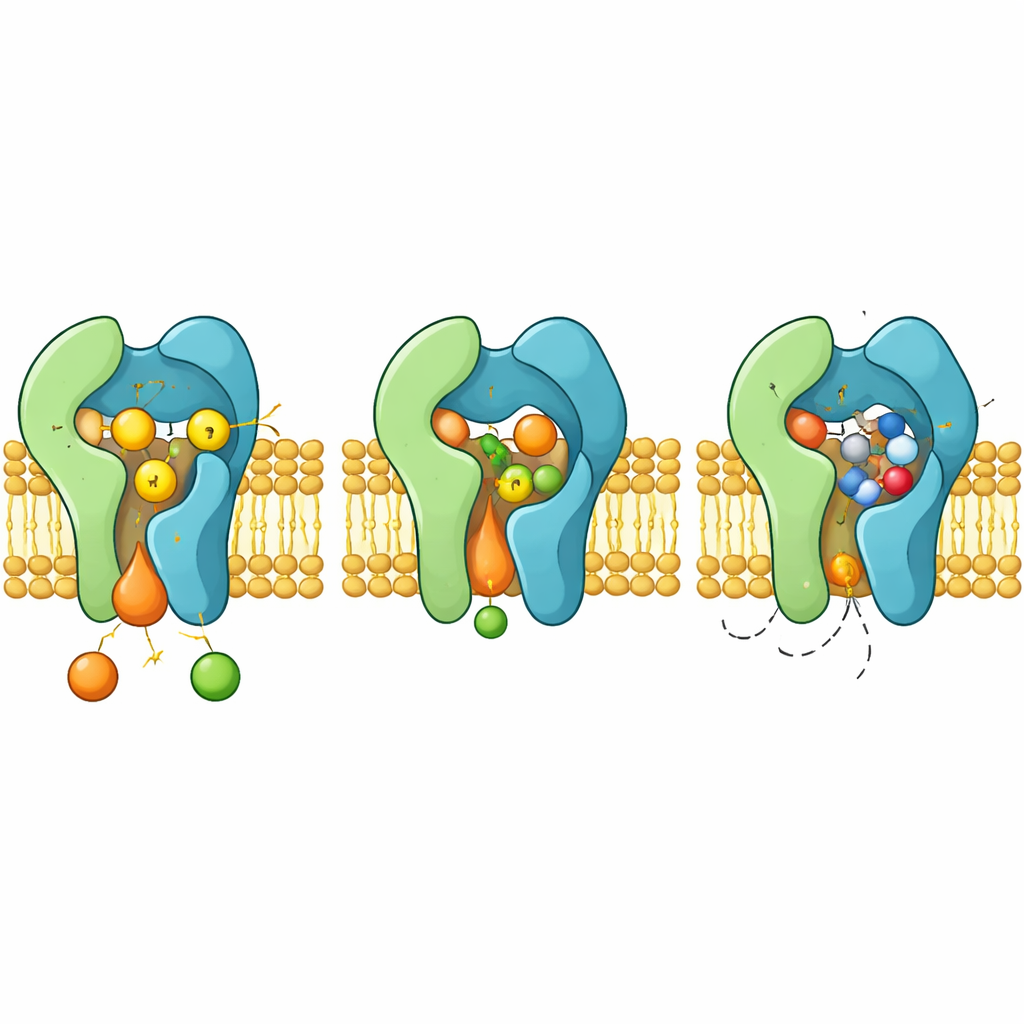
How the drug blocks and how resistance arises
When aureobasidin A binds, it occupies the same reaction chamber that normally hosts ceramide. The cyclic molecule clamps into place between flexible loops and helices, pushing aside aromatic side chains that reorient to create a snug pocket. In this configuration, the drug occludes the H–H–D triad and sterically excludes ceramide, acting as a classic competitive inhibitor. The structures also explain how specific mutations in Aur1, such as altering residues H157 or F158 near the pocket, weaken drug binding while preserving catalysis, giving rise to resistant fungi.
What this means for future antifungal drugs
Together, these findings provide a detailed map of an enzyme that fungi cannot live without and humans do not possess. By showing exactly how IPC synthase assembles, how it recognizes its lipid substrates, and how aureobasidin A wedges into its reaction chamber, the study offers a blueprint for designing new drugs that selectively cripple fungal membranes. Such structure-guided compounds could expand the limited antifungal arsenal and help outpace the growing problem of drug‑resistant fungal infections.
Citation: Chen, J., Ke, Y., Zhang, M. et al. Molecular insights into fungal inositol phosphorylceramide synthesis and its inhibition by antifungal aureobasidin A. Nat Commun 17, 3951 (2026). https://doi.org/10.1038/s41467-026-69777-3
Keywords: antifungal drugs, fungal cell membrane, sphingolipids, drug resistance, cryo electron microscopy