Clear Sky Science · es
Perspectivas moleculares sobre la síntesis fúngica de inositol fosforilceramida y su inhibición por el antifúngico aureobasidina A
Por qué importa esta historia sobre hongos
Las infecciones fúngicas invasivas causan silenciosamente millones de muertes cada año, sobre todo en personas con el sistema inmunitario debilitado. Sin embargo, los médicos cuentan con solo unos pocos fármacos antifúngicos y muchos hongos están adquiriendo resistencia. Este estudio se centra en un proceso fúngico que no existe en las células humanas, lo que lo convierte en un blanco ideal para nuevos medicamentos. Al desvelar cómo funciona una enzima fúngica clave —y cómo un fármaco potente la bloquea—, los investigadores sientan las bases para terapias antifúngicas más seguras y eficaces.
Un punto débil único en la armadura fúngica
Las células fúngicas dependen de una clase especial de lípidos, llamados esfingolípidos, para construir y organizar sus membranas. Uno de ellos, la inositol fosforilceramida (IPC), se encuentra en los hongos pero está ausente en los mamíferos. La IPC se sintetiza mediante un complejo enzimático de dos componentes conocido como IPC sintasa. Un socio, Aur1, realiza la química; el otro, Kei1, ayuda a posicionar y estabilizar el complejo en la membrana. Dado que los hongos no pueden vivir sin la IPC sintasa, mientras que los humanos no la poseen, esta enzima ha sido considerada durante mucho tiempo un objetivo farmacológico atractivo.
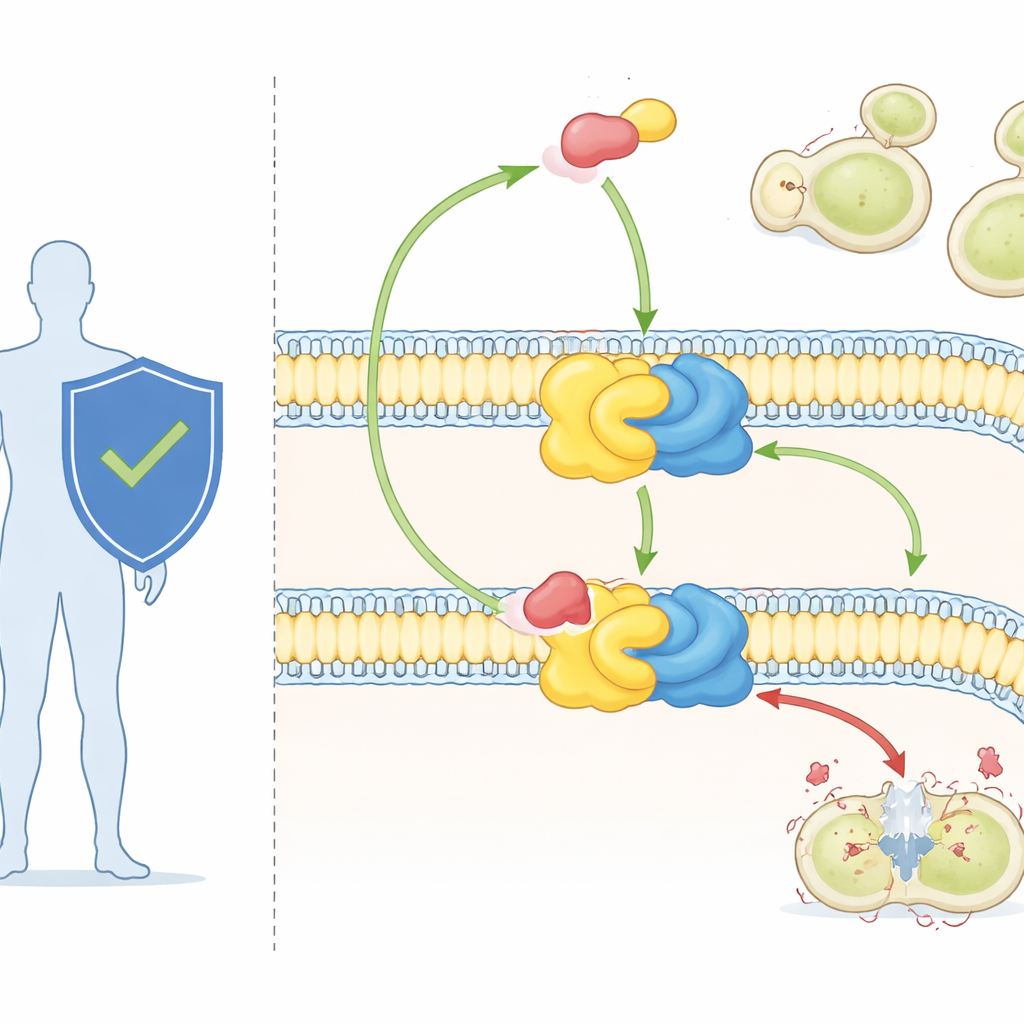
Un antifúngico potente y un blanco enigmático
La aureobasidina A, usada hasta ahora principalmente en investigación, es uno de los antifúngicos más potentes conocidos. Puede matar patógenos fúngicos importantes a dosis muy bajas y muestra escasa toxicidad para células de mamíferos. Estudios genéticos ya habían demostrado que cambios en la proteína Aur1 pueden conferir resistencia al fármaco, confirmando que la IPC sintasa es su diana. Pero sin una vista de alta resolución de la enzima, los científicos no podían ver con precisión cómo reconoce sus sustratos lipídicos naturales ni cómo la aureobasidina A se inserta para paralizar la reacción.
Congelar la enzima en acción
Para resolver este problema, los investigadores utilizaron crio‑microscopía electrónica, una técnica que imagen moléculas flash‑congeladas con detalle cercano al atómico. La IPC sintasa es pequeña y en su mayor parte está enterrada en la membrana, lo que dificulta su visualización. El equipo superó esto añadiendo una etiqueta proteica diminuta y un nanocuerpo a modo de “asa” que mejoró la alineación de las imágenes sin alterar la actividad de la enzima ni su sensibilidad al fármaco. A continuación determinaron las estructuras de la IPC sintasa de levadura en dos estados clave: unida a su sustrato lipídico ceramida y unida a la aureobasidina A.
La cavidad de trabajo de la enzima y su socio de control
Las estructuras revelan que Aur1 y Kei1 forman una pareja compacta que atraviesa la membrana. Aur1 contiene una cámara de reacción en forma de ranura que se abre lateralmente hacia los lípidos circundantes. La ceramida se aloja en esa ranura, con una de sus colas enterrada profundamente a lo largo de una línea de aminoácidos hidrofóbicos y su cabeza reactiva orientada hacia tres residuos cruciales —dos histidinas y un aspartato— situados en la base de la cámara. Mutar cualquiera de los miembros de esta "tríada H–H–D" abolía por completo la actividad enzimática, lo que demuestra que juntos llevan a cabo la transferencia en dos pasos de un grupo portador de fosfato de un lípido a otro.
Kei1, que se consideraba antes un ayudante pasivo, resulta ser esencial. Establece contactos extensos con un bucle de Aur1 y envuelve un bolsillo compartido que une firmemente una molécula de fosfolípido. Cuando se mutan residuos clave que fijan este lípido, el ensamblaje Aur1–Kei1 se desintegra y la actividad se pierde. Esto sugiere que los lípidos nativos de la membrana actúan como cuñas estructurales que estabilizan el complejo enzimático, indicando maneras adicionales en que los fármacos podrían perturbar su función.
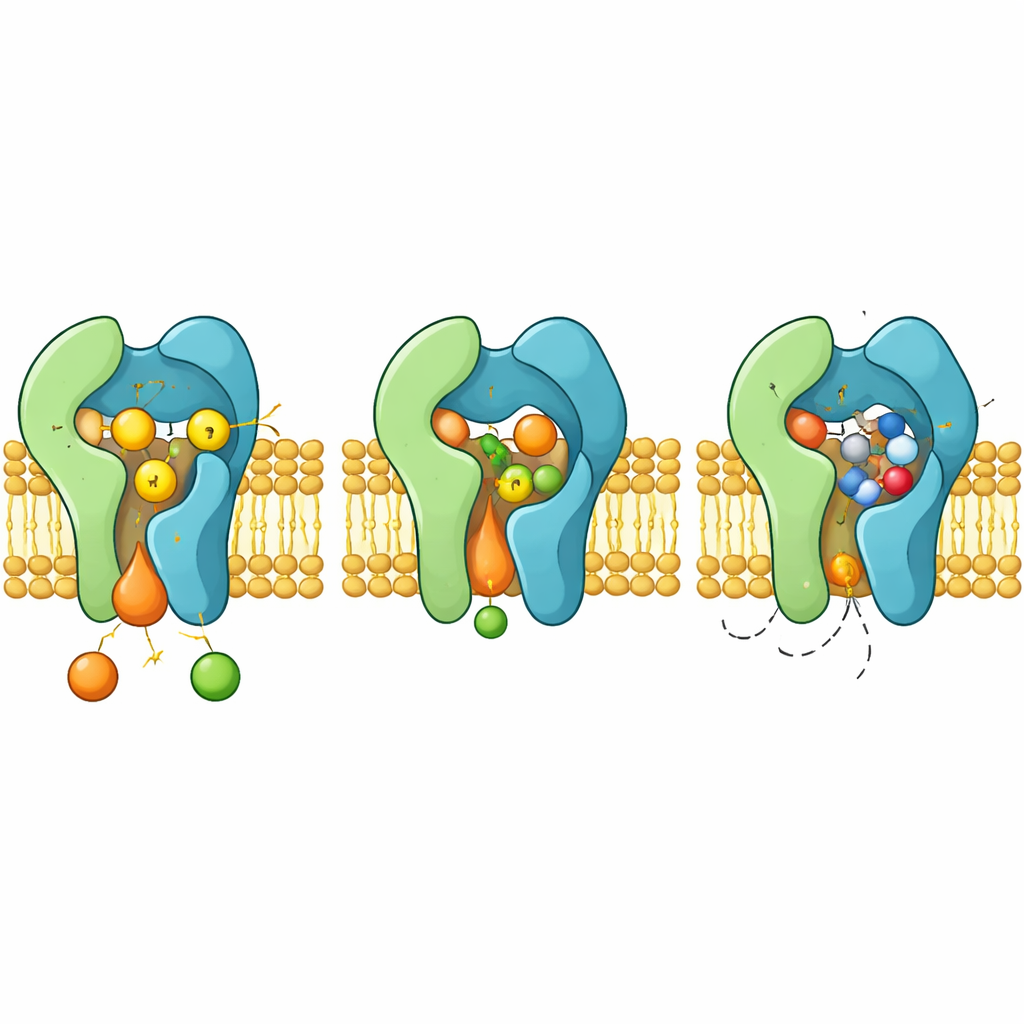
Cómo bloquea el fármaco y cómo aparece la resistencia
Cuando la aureobasidina A se une, ocupa la misma cámara de reacción que normalmente aloja la ceramida. La molécula cíclica se fija entre bucles y hélices flexibles, desplazando cadenas aromáticas que se reorientan para crear un bolsillo ajustado. En esta configuración, el fármaco tapa la tríada H–H–D e impide estéricamente la entrada de la ceramida, actuando como un inhibidor competitivo clásico. Las estructuras también explican cómo mutaciones específicas en Aur1, como la modificación de los residuos H157 o F158 cerca del bolsillo, debilitan la unión del fármaco mientras preservan la catálisis, provocando la aparición de hongos resistentes.
Qué significa esto para futuros antifúngicos
En conjunto, estos hallazgos proporcionan un mapa detallado de una enzima indispensable para los hongos y ausente en los humanos. Al mostrar exactamente cómo se ensambla la IPC sintasa, cómo reconoce sus sustratos lipídicos y cómo la aureobasidina A se incrusta en su cámara de reacción, el estudio ofrece un plano para diseñar nuevos fármacos que incapaciten selectivamente las membranas fúngicas. Compuestos guiados por la estructura podrían ampliar el limitado arsenal antifúngico y ayudar a adelantarse al creciente problema de las infecciones fúngicas resistentes a fármacos.
Cita: Chen, J., Ke, Y., Zhang, M. et al. Molecular insights into fungal inositol phosphorylceramide synthesis and its inhibition by antifungal aureobasidin A. Nat Commun 17, 3951 (2026). https://doi.org/10.1038/s41467-026-69777-3
Palabras clave: fármacos antifúngicos, membrana celular fúngica, esfingolípidos, resistencia a fármacos, crio‑microscopía electrónica