Clear Sky Science · de
Molekulare Einblicke in die Synthese von inositolphosphorylceramid bei Pilzen und deren Hemmung durch das Antimykotikum Aureobasidin A
Warum diese Pilzgeschichte wichtig ist
Invasive Pilzinfektionen fordern weltweit stillschweigend Millionen von Todesfällen pro Jahr, insbesondere bei Menschen mit geschwächtem Immunsystem. Ärztinnen und Ärzte verfügen jedoch nur über eine kleine Auswahl an Antimykotika, und viele Pilze entwickeln zunehmend Resistenzen. Diese Studie konzentriert sich auf einen pilzspezifischen Prozess, der in menschlichen Zellen nicht vorkommt und daher ein ideales Ziel für neue Wirkstoffe darstellt. Indem die Forschenden aufdecken, wie ein zentrales pilzliches Enzym arbeitet — und wie ein starkes Mittel es blockiert —, schaffen sie die Grundlage für sicherere, wirksamere Antimykotika.
Eine einzigartige Schwachstelle in der pilzlichen Rüstung
Pilzzellen stützen sich auf eine spezielle Klasse von Fetten, sogenannte Sphingolipide, um ihre Membranen aufzubauen und zu organisieren. Eines davon, inositolphosphorylceramid (IPC), kommt bei Pilzen vor, nicht aber bei Säugetieren. IPC wird von einem zweiteiligen Enzymkomplex gebildet, der als IPC-Synthase bekannt ist. Ein Part, Aur1, führt die chemische Reaktion aus; der andere, Kei1, hilft dabei, den Komplex in der Membran zu positionieren und zu stabilisieren. Da Pilze ohne IPC-Synthase nicht lebensfähig sind und Menschen dieses Enzym überhaupt nicht besitzen, gilt es schon lange als attraktives Wirkstoffziel.
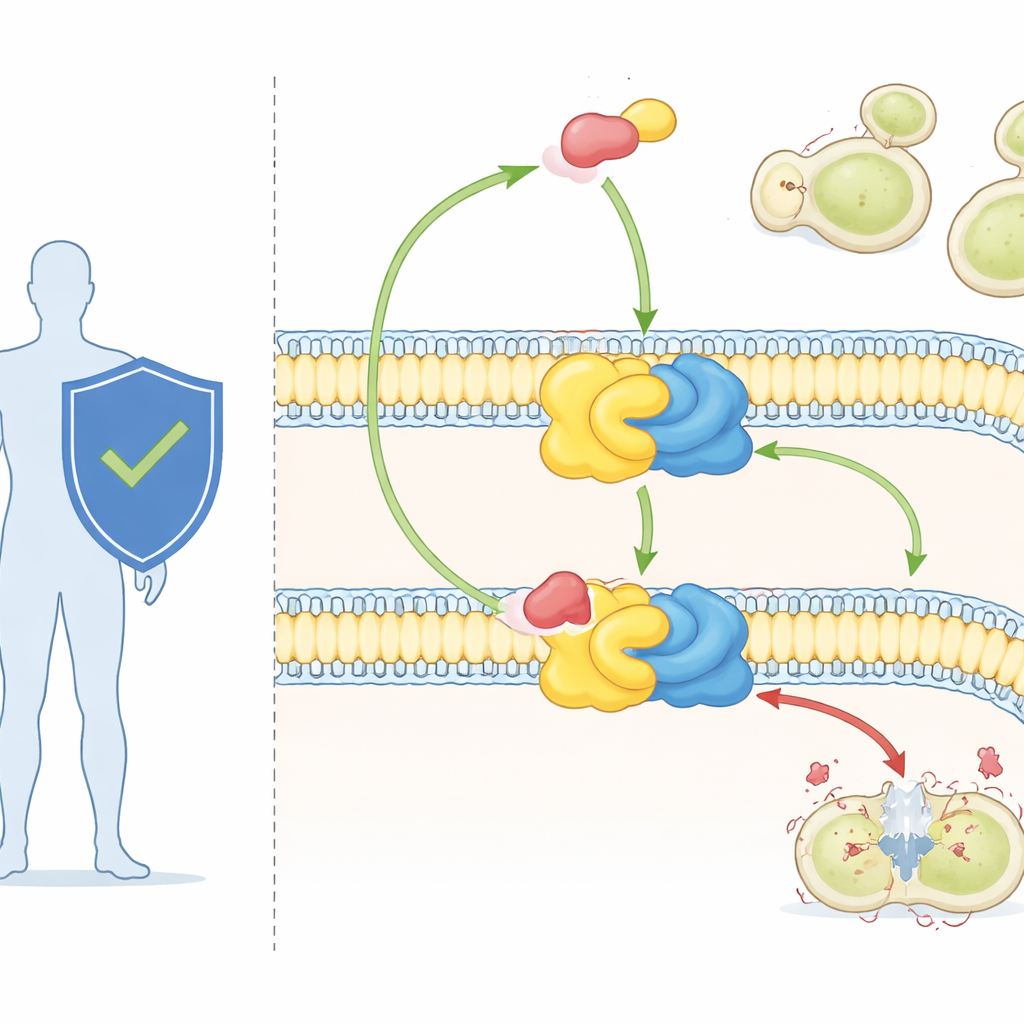
Ein starkes Antimykotikum und ein rätselhaftes Ziel
Das Medikament Aureobasidin A, bislang vor allem in der Forschung eingesetzt, gehört zu den stärksten bekannten Antimykotika. Es kann wichtige Krankheitserreger der Pilze in sehr geringen Dosen abtöten und zeigt nur geringe Toxizität gegenüber Säugetierzellen. Genetische Arbeiten hatten bereits gezeigt, dass Veränderungen im Aur1-Protein Pilze resistent gegen das Mittel machen können, was bestätigt, dass die IPC-Synthase sein Ziel ist. Ohne eine hochaufgelöste Ansicht des Enzyms konnten Wissenschaftlerinnen und Wissenschaftler jedoch nicht genau sehen, wie es seine natürlichen Lipid-Substrate erkennt oder wie Aureobasidin A sich so einfügt, dass die Reaktion gestoppt wird.
Das Enzym in Aktion einfrieren
Um dieses Problem zu lösen, nutzte das Team kryo‑Elektronenmikroskopie, eine Technik, die schockgefrorene Moleküle nahezu auf atomarer Ebene abbildet. Die IPC-Synthase ist klein und größtenteils in der Membran verborgen, was ihre Darstellung erschwert. Die Forschenden überwandenen dies, indem sie ein winziges Protein‑Tag und eine passende Nanobody‑„Greifer“‑Bindung anfügten, die die Bildausrichtung verbesserten, ohne die Aktivität des Enzyms oder seine Empfindlichkeit gegenüber dem Wirkstoff zu stören. Anschließend bestimmten sie Strukturen der Hefe‑IPC‑Synthase in zwei zentralen Zuständen: gebunden an ihr Lipid‑Substrat Ceramid und gebunden an Aureobasidin A.
Die Arbeitskammer des Enzyms und der Kontrollpartner
Die Strukturen zeigen, dass Aur1 und Kei1 ein enges Paar bilden, das die Membran überspannt. Aur1 enthält eine rillenartige Reaktionskammer, die seitlich in die umgebenden Lipide öffnet. Ceramid lagert sich in dieser Rille ein, wobei ein Tail tief entlang einer Reihe öliger Aminosäuren vergraben ist und der reaktive Kopf auf drei entscheidende Reste — zwei Histidine und eine Asparaginsäure — zeigt, die am Boden der Kammer sitzen. Mutationen eines jeden Mitglieds dieser „H–H–D‑Triade“ beseitigen die Enzymaktivität vollständig, was zeigt, dass sie gemeinsam den zweistufigen Transfer einer phosphatehaltigen Gruppe von einem Lipid auf ein anderes ausführen.
Kei1, einst als passiver Helfer angesehen, erweist sich als essentiell. Es bildet ausgedehnte Kontakte mit einer Schleife von Aur1 und umschließt eine gemeinsame Tasche, die ein Phospholipidmolekül fest bindet. Werden Schlüsselreste, die dieses Lipid an Ort und Stelle halten, mutiert, fällt der Aur1–Kei1‑Zusammenbau auseinander und die Aktivität geht verloren. Das legt nahe, dass native Membranlipide als strukturelle Keile wirken, die den Enzymkomplex stabilisieren, und weist auf zusätzliche Möglichkeiten hin, wie Wirkstoffe seine Funktion stören könnten.
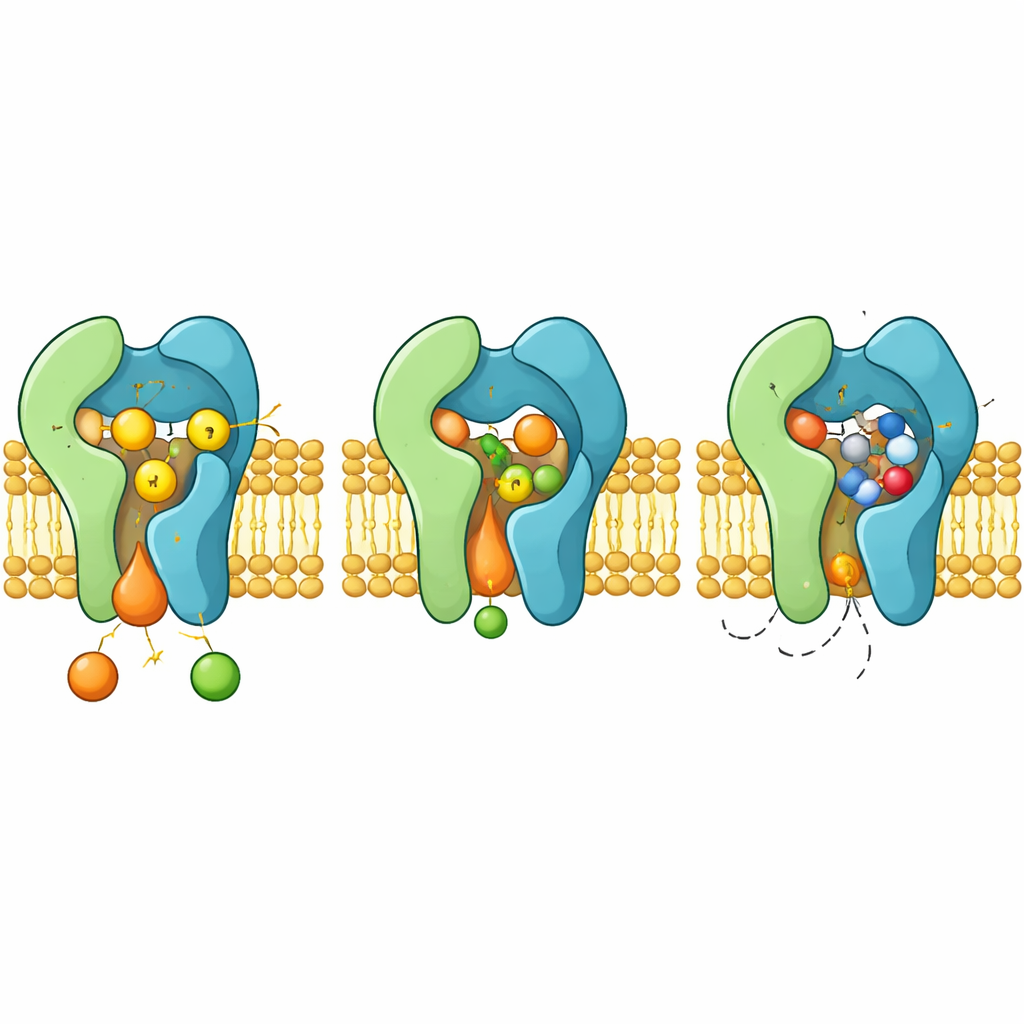
Wie das Medikament blockiert und wie Resistenz entsteht
Wenn Aureobasidin A bindet, besetzt es dieselbe Reaktionskammer, die normalerweise Ceramid beherbergt. Das zyklische Molekül klemmt sich zwischen flexible Schleifen und Helices und verdrängt aromatische Seitengruppen, die sich neu orientieren, um eine enge Tasche zu schaffen. In dieser Konfiguration schirmt das Medikament die H–H–D‑Triade ab und schließt Ceramid sterisch aus, wirkt also als klassischer kompetitiver Inhibitor. Die Strukturen erklären auch, wie bestimmte Mutationen in Aur1, etwa Veränderungen der Reste H157 oder F158 in der Nähe der Tasche, die Medikamentbindung schwächen, während die Katalyse erhalten bleibt — was zur Entstehung resistenter Pilzstämme führt.
Was das für zukünftige Antimykotika bedeutet
Zusammen liefern diese Befunde eine detaillierte Landkarte eines Enzyms, das Pilze zum Überleben brauchen und das beim Menschen nicht vorhanden ist. Indem die Studie zeigt, wie sich die IPC‑Synthase zusammensetzt, wie sie ihre Lipid‑Substrate erkennt und wie Aureobasidin A in ihre Reaktionskammer einschiebt, bietet sie einen Bauplan zur Entwicklung neuer Wirkstoffe, die gezielt die pilzliche Membran angreifen. Solche strukturgeleiteten Verbindungen könnten das begrenzte Arsenal an Antimykotika erweitern und dazu beitragen, dem wachsenden Problem medikamentenresistenter Pilzinfektionen voraus zu sein.
Zitation: Chen, J., Ke, Y., Zhang, M. et al. Molecular insights into fungal inositol phosphorylceramide synthesis and its inhibition by antifungal aureobasidin A. Nat Commun 17, 3951 (2026). https://doi.org/10.1038/s41467-026-69777-3
Schlüsselwörter: antimykotische Medikamente, pilzliche Zellmembran, sphingolipide, Medikamentenresistenz, kryoelektronenmikroskopie