Clear Sky Science · it
Prospettive molecolari sulla sintesi fungina di inositol fosforilceramide e la sua inibizione dall'antimicotico aureobasidin A
Perché questa storia fungina è importante
Le infezioni fungine invasive uccidono silenziosamente milioni di persone ogni anno, soprattutto chi ha un sistema immunitario indebolito. Eppure i medici dispongono di pochi farmaci antifungini, e molti funghi stanno evolvendo resistenze. Questo studio si concentra su un processo fungino assente nelle cellule umane, rendendolo un bersaglio ideale per nuove terapie. Scoprendo come funziona un enzima fungino chiave — e come un potente farmaco lo blocca — i ricercatori gettano le basi per terapie antifungine più sicure ed efficaci.
Un punto debole unico nell'armatura fungina
Le cellule fungine si affidano a una classe speciale di lipidi, chiamati sfingolipidi, per costruire e organizzare le loro membrane. Uno di questi, l'inositol fosforilceramide (IPC), si trova nei funghi ma è assente nei mammiferi. L'IPC è prodotto da un complesso enzimatico composto da due parti noto come IPC sintetasi. Un partner, Aur1, esegue la reazione chimica; l'altro, Kei1, aiuta a posizionare e stabilizzare il complesso nella membrana. Poiché i funghi non possono vivere senza l'IPC sintetasi, mentre gli esseri umani non la possiedono affatto, questo enzima è stato a lungo considerato un bersaglio farmacologico attraente.
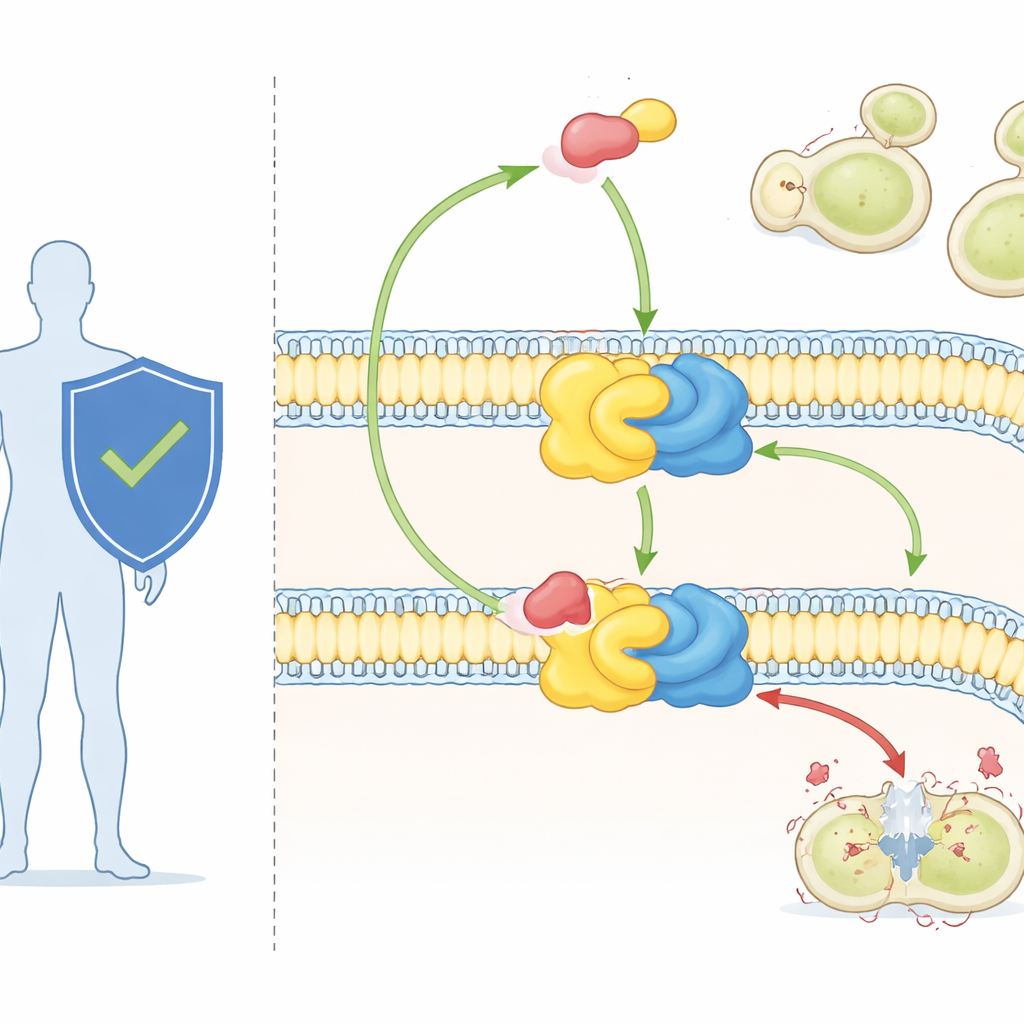
Un antifungino potente e un bersaglio misterioso
Il composto aureobasidin A, usato finora soprattutto nella ricerca, è uno degli antifungini più potenti conosciuti. Può uccidere patogeni fungini importanti a dosi piccolissime e mostra scarsa tossicità per le cellule dei mammiferi. Dati genetici avevano già dimostrato che alterazioni della proteina Aur1 possono rendere i funghi resistenti al farmaco, confermando che l'IPC sintetasi è il suo bersaglio. Ma senza una visione ad alta risoluzione dell'enzima, gli scienziati non potevano vedere esattamente come riconosca i suoi substrati lipidici naturali né come l'aureobasidin A si infili per bloccare la reazione.
Congelare l'enzima in azione
Per risolvere questo problema, i ricercatori hanno utilizzato la criomicroscopia elettronica, una tecnica che visualizza molecole rapidamente congelate a dettaglio quasi atomico. L'IPC sintetasi è piccola e per lo più sepolta nella membrana, il che la rende difficile da visualizzare. Il team ha superato questa difficoltà attaccando un piccolo tag proteico e un nanobody abbinato come “maniglia” che ha migliorato l'allineamento delle immagini senza alterare l'attività dell'enzima né la sua sensibilità al farmaco. Hanno quindi determinato le strutture della IPC sintetasi di lievito in due stati chiave: legata al suo substrato lipidico ceramide e legata all'aureobasidin A.
La cavità funzionale dell'enzima e il partner di controllo
Le strutture rivelano Aur1 e Kei1 che formano una coppia stretta che attraversa la membrana. Aur1 contiene una camera di reazione a forma di solco che si apre lateralmente verso i lipidi circostanti. La ceramide si annida in questo solco, con una delle code sepolta in profondità lungo una linea di residui aminoacidici apolari e la testa reattiva rivolta verso tre residui cruciali — due istidine e un aspartato — che si trovano alla base della camera. La mutazione di qualsiasi membro di questa “triade H–H–D” annulla completamente l'attività enzimatica, dimostrando che insieme eseguono il trasferimento in due fasi di un gruppo fosforilato da un lipide a un altro.
Kei1, un tempo ritenuto un semplice aiuto passivo, si rivela essenziale. Stabilisce contatti estesi con una spira di Aur1 e abbraccia una tasca condivisa che lega saldamente una molecola di fosfolipide. Quando residui chiave che tengono questo lipide in posizione vengono mutati, l'assemblaggio Aur1–Kei1 si disfa e l'attività viene persa. Ciò suggerisce che i lipidi nativi della membrana agiscano come cunei strutturali che stabilizzano il complesso enzimatico, indicando modalità aggiuntive con cui i farmaci potrebbero disturbare la sua funzione.
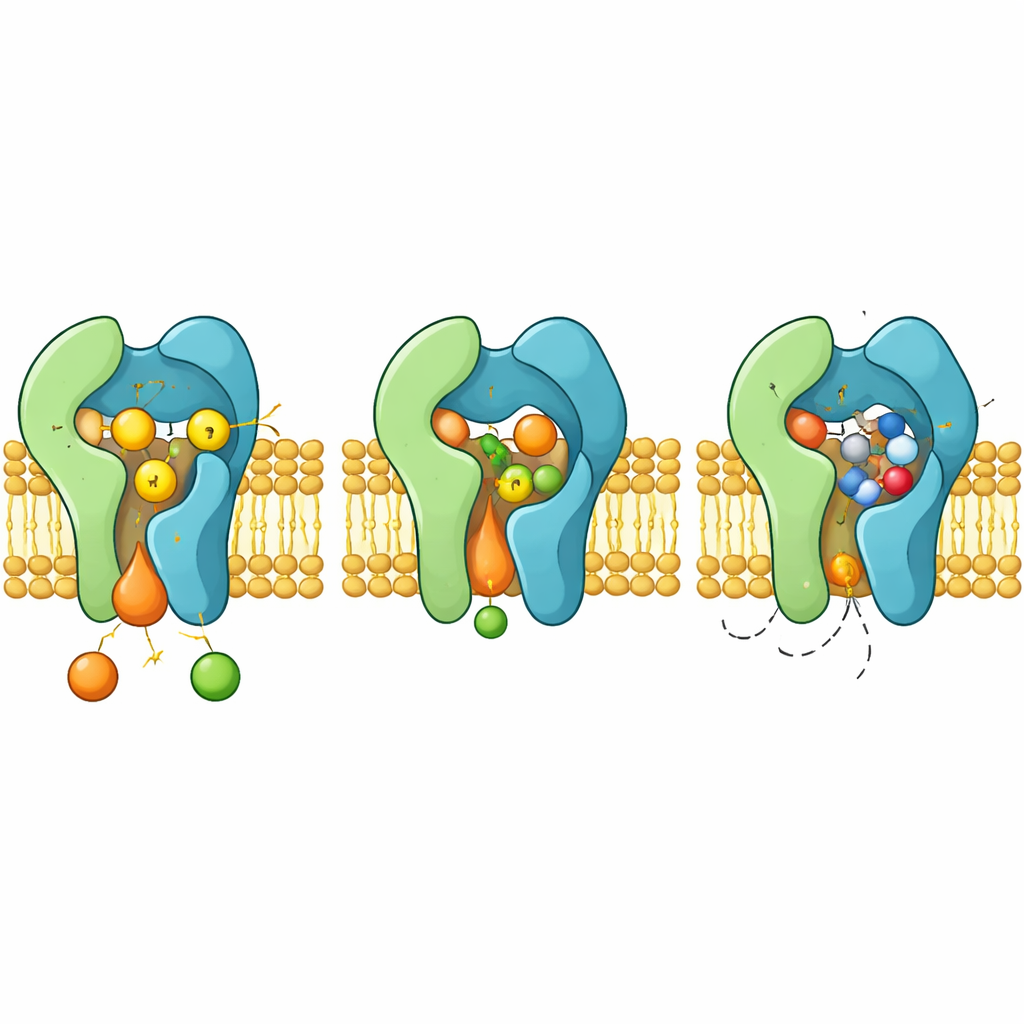
Come il farmaco blocca e come emerge la resistenza
Quando l'aureobasidin A si lega, occupa la stessa camera di reazione che normalmente ospita la ceramide. La molecola ciclica si blocca tra anse e eliche flessibili, spingendo da parte catene laterali aromatiche che si riorientano per creare una tasca aderente. In questa configurazione, il farmaco occlude la triade H–H–D ed esclude stericamente la ceramide, agendo come un classico inibitore competitivo. Le strutture spiegano anche come specifiche mutazioni in Aur1, come l'alterazione dei residui H157 o F158 vicino alla tasca, indeboliscano il legame con il farmaco pur preservando la catalisi, dando origine a funghi resistenti.
Cosa significa per i futuri farmaci antifungini
Nel loro insieme, questi risultati forniscono una mappa dettagliata di un enzima di cui i funghi non possono fare a meno e che gli esseri umani non possiedono. Mostrando esattamente come si assembla l'IPC sintetasi, come riconosce i suoi substrati lipidici e come l'aureobasidin A si incastra nella sua camera di reazione, lo studio offre un progetto guida per ideare nuovi farmaci che compromettano selettivamente le membrane fungine. Composti progettati sulla base di queste strutture potrebbero ampliare l'arsenale antifungino limitato e contribuire a contrastare il crescente problema delle infezioni fungine resistenti ai farmaci.
Citazione: Chen, J., Ke, Y., Zhang, M. et al. Molecular insights into fungal inositol phosphorylceramide synthesis and its inhibition by antifungal aureobasidin A. Nat Commun 17, 3951 (2026). https://doi.org/10.1038/s41467-026-69777-3
Parole chiave: farmaci antifungini, membrana cellulare fungina, sfingolipidi, resistenza ai farmaci, criomicroscopia elettronica