Clear Sky Science · pt
Sinalização de cálcio anômala e atividade neuronal no modelo iPSC L271H CACNA1D (Cav1.3) de doenças do neurodesenvolvimento
Quando uma única mudança gênica reverbera pelo desenvolvimento cerebral
Mutações raras em genes podem alterar sutilmente como as células cerebrais lidam com sinais elétricos e químicos. Este estudo acompanha uma dessas alterações em um gene de canal de cálcio associado a atraso no desenvolvimento, convulsões e sintomas semelhantes ao autismo. Ao transformar células sanguíneas de uma paciente em células-tronco e depois em tecidos cerebrais e neurônios em miniatura em cultura, os pesquisadores mapeiam como um ajuste molecular minúsculo pode distorcer o desenvolvimento cerebral precoce e a atividade neuronal.
Guardiões que deixam o cálcio entrar nas células cerebrais
As células cerebrais dependem de um fluxo controlado de íons cálcio para crescer, comunicar-se e ligar ou desligar genes. “Portas” especializadas na membrana celular, chamadas canais de cálcio dependentes de voltagem, abrem durante a atividade elétrica e permitem a entrada de cálcio. Um desses canais, conhecido como Cav1.3, é importante em muitos órgãos, incluindo o cérebro e tecidos produtores de hormônios. Em vários pacientes, alterações raras herdadas no gene Cav1.3 têm sido associadas a um conjunto de sintomas: desequilíbrios hormonais, fraqueza muscular, convulsões, desenvolvimento tardio e traços do espectro autista. A variante estudada aqui, chamada L271H, havia mostrado em células não neuronais que torna o canal mais fácil de abrir, mas seu impacto em células cerebrais humanas era desconhecido.
Construindo células cerebrais específicas do paciente no laboratório
A equipe criou um modelo iPSC, no qual células sanguíneas de uma menina portadora da variante L271H foram reprogramadas em células-tronco capazes de formar qualquer tecido. Essas células-tronco pluripotentes induzidas foram então direcionadas para se tornarem progenitores neurais — os blocos de construção iniciais que depois geram neurônios — e neurônios do mesencéfalo relacionados ao sinal de dopamina. Os pesquisadores também cultivaram organoides corticais tridimensionais, pequenos “mini-cérebros” que imitam estágios iniciais da formação do córtex humano. Ao comparar células derivadas da paciente com várias linhas controle de doadores saudáveis, puderam separar os efeitos da variante da variação genética natural.
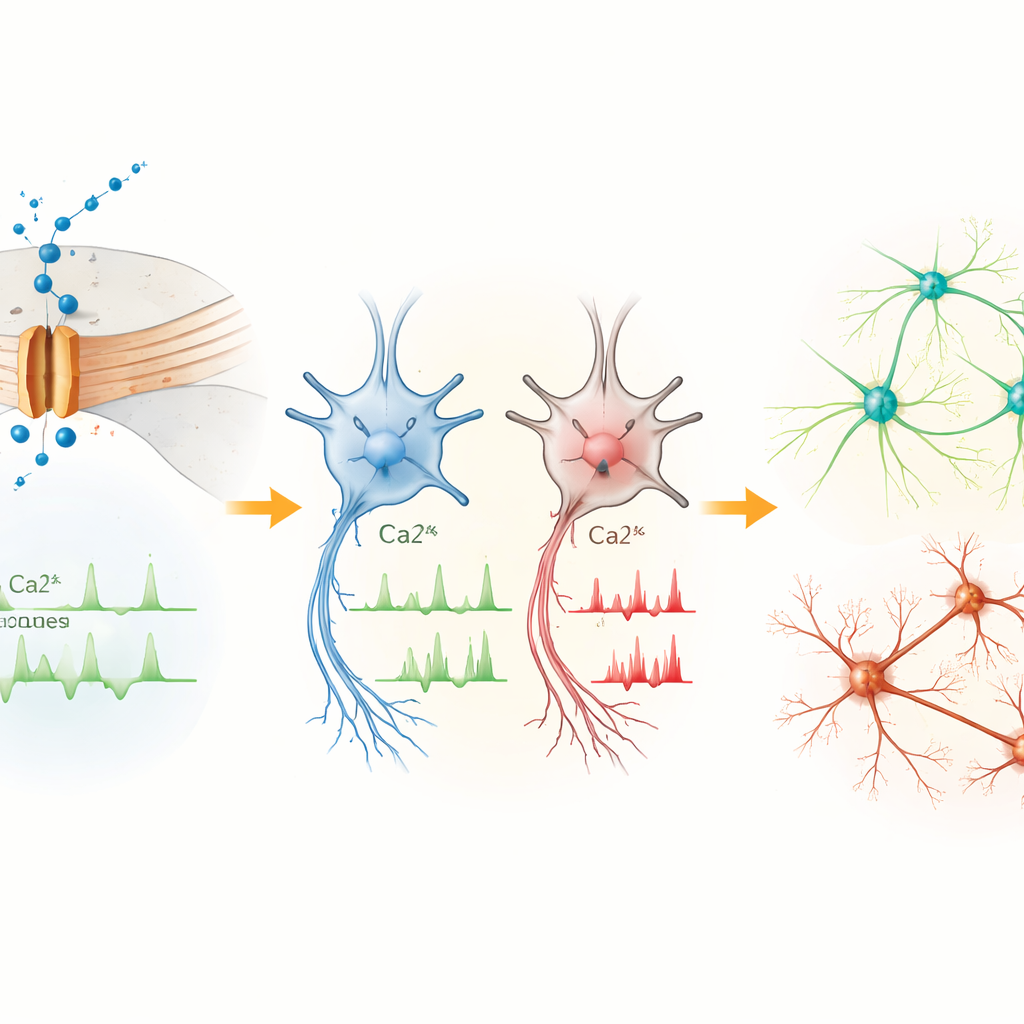
As células precoces sinalizam demais, neurônios maduros disparam de menos
Nos progenitores neurais mais precoces, o canal Cav1.3 foi a forma dominante desse portão de cálcio. Células portadoras da variante L271H apresentaram surtos espontâneos de cálcio mais frequentes do que os controles, embora suas propriedades elétricas básicas fossem semelhantes. Isso sugere que, nessa fase, a variante perturba principalmente o equilíbrio de cálcio intracelular em vez de causar disparos elétricos ostensivos. À medida que as células amadureceram em neurônios do mesencéfalo, o quadro se inverteu. Embora outro tipo de canal de cálcio tenha se tornado mais abundante no geral, neurônios com a variante L271H mostraram menos picos de cálcio, um estado de repouso mais despolarizado e foram mais difíceis de induzir a disparos repetidos. Seus potenciais de ação foram menores e mais lentos para subir e cair, indicando que a variante, em última análise, atenua a capacidade dos neurônios de enviar sinais elétricos robustos.
Mini-cérebros que crescem mais rápido mas se organizam mal
Nos organoides corticais, a variante L271H deixou uma marca forte na estrutura. Em comparação com os controles, os organoides derivados da paciente tinham menos cavidades semelhantes a ventrículos e de menor tamanho — zonas em forma de anel onde os progenitores normalmente se alinham de forma ordenada. Células da glia radial, que atuam como andaimes e guiam os neurônios recém-formados, estavam dispersas por todo o tecido em vez de formarem camadas polarizadas e organizadas. Progenitores intermediários e neurônios surgiram mais cedo e depois diminuíram mais cedo, sugerindo que os progenitores foram impulsionados a se diferenciar rápido demais. Aos 60 dias, o número total de neurônios se igualou, mas o descompasso temporal inicial e a desorganização apontam para uma planta baixa alterada para a construção de circuitos corticais, em vez de uma falha absoluta em produzir neurônios.
Atividade gênica deslocada para programas de risco do neurodesenvolvimento
Para entender como o manejo alterado do cálcio impacta o controle gênico, a equipe sequenciou RNA de células da paciente e de controles em diferentes estágios. As diferenças foram modestas nas células-tronco, mas cresceram nos progenitores e neurônios à medida que a expressão de Cav1.3 aumentou. Muitos genes regulados para cima ou para baixo nas células da paciente estão envolvidos no controle da transcrição, desenvolvimento cerebral e neurogênese. Vários, incluindo PTN, MEIS2, POU3F2, CNTN4, CNTNAP2 e AUTS2, são genes de risco conhecidos para autismo e condições do neurodesenvolvimento relacionadas. Essas alterações sugerem que a entrada anômala de cálcio através de Cav1.3 remodela programas gênicos dependentes de atividade que orientam como as células cerebrais proliferam, migram e se conectam.
O que isso significa para pacientes e terapias futuras
Em termos simples, este trabalho mostra como uma única variante num canal de cálcio pode primeiro superestimar células cerebrais precoces, depois deixar neurônios maduros subpotenciados, ao mesmo tempo em que desloca redes gênicas associadas a transtornos do neurodesenvolvimento. O resultado é um cérebro que pode parecer amplamente normal externamente, mas é construído a partir de circuitos que se desenvolveram rápido demais e se organizaram de forma imperfeita. Ao recriar a mutação desta paciente em células e organoides humanos, o estudo fornece um mapa detalhado que liga um defeito molecular a sinalização celular alterada, arquitetura tecidual e regulação gênica. Esse modelo agora oferece uma plataforma para testar drogas direcionadas e estratégias genéticas com o objetivo de restaurar uma sinalização de cálcio mais próxima do normal em canalopatias relacionadas à Cav1.3.
Citação: Tisch, M., Geisler, S.M., Gabassi, E. et al. Aberrant calcium signaling and neuronal activity in the L271H CACNA1D (Cav1.3) iPSC model of neurodevelopmental disease. Mol Psychiatry 31, 2927–2940 (2026). https://doi.org/10.1038/s41380-025-03429-8
Palavras-chave: canais de cálcio, transtornos do neurodesenvolvimento, células-tronco pluripotentes induzidas, organoides cerebrais, genes de risco para autismo