Clear Sky Science · fr
Signalisation calcique aberrante et activité neuronale dans le modèle iPSC L271H CACNA1D (Cav1.3) des maladies neurodéveloppementales
Quand une simple modification génétique affecte le développement cérébral
Des variants rares dans des gènes peuvent modifier subtilement la façon dont les cellules cérébrales gèrent les signaux électriques et chimiques. Cette étude suit l’un de ces variants dans un gène de canal calcique associé à un retard de développement, à des crises et à des symptômes proches de l’autisme. En reprogrammant les cellules sanguines d’une patiente en cellules souches puis en tissus cérébraux miniatures et en neurones en culture, les chercheurs cartographient comment une infime altération moléculaire peut déformer le développement précoce du cerveau et l’activité neuronale.
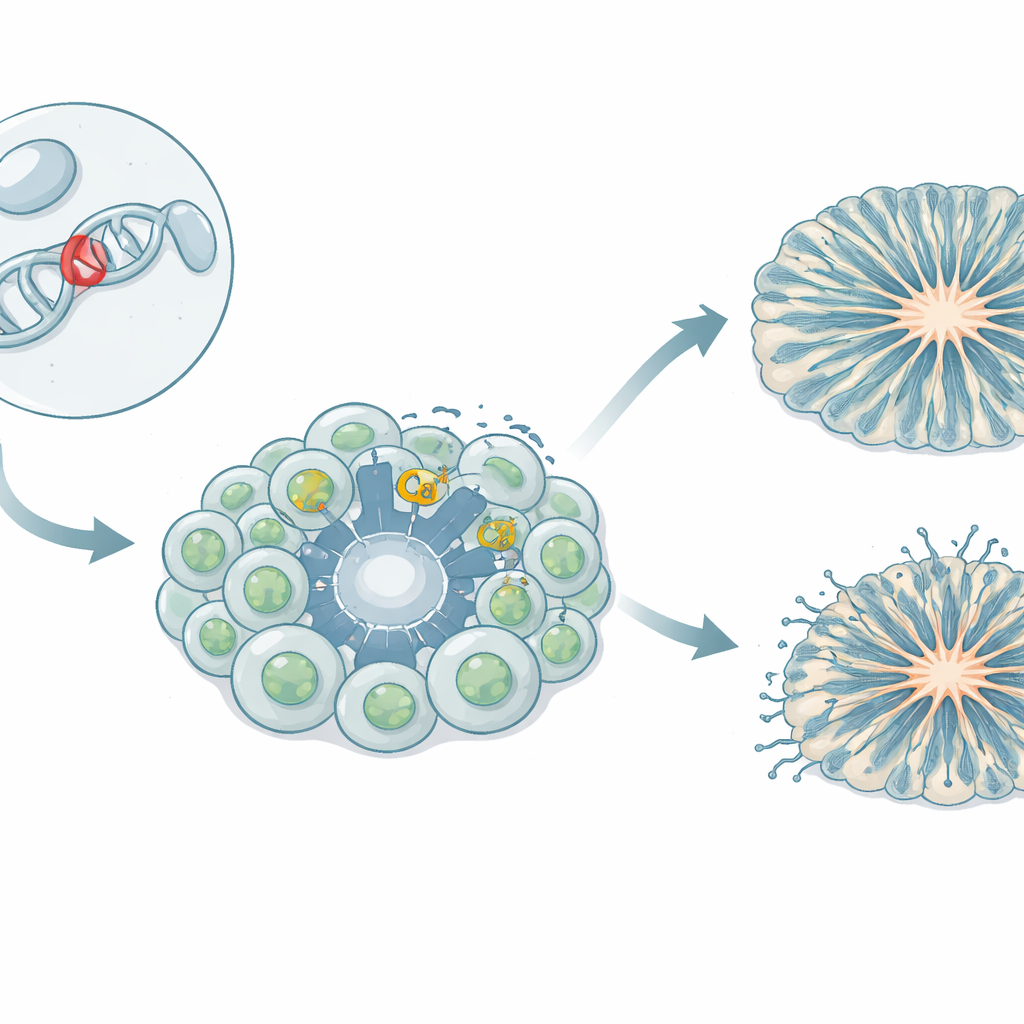
Des portiers qui laissent entrer le calcium dans les cellules cérébrales
Les cellules cérébrales dépendent d’un flux contrôlé d’ions calcium pour croître, communiquer entre elles et activer ou désactiver certains gènes. Des « portes » spécialisées dans la membrane cellulaire, appelées canaux calciques voltage‑dépendants, s’ouvrent lors de l’activité électrique et laissent entrer le calcium. L’un de ces canaux, nommé Cav1.3, est important dans de nombreux organes, y compris le cerveau et les tissus producteurs d’hormones. Chez plusieurs patients, des variants rares hérités du gène Cav1.3 ont été associés à un ensemble de symptômes : déséquilibres hormonaux, faiblesse musculaire, crises, retard du développement et traits du spectre autistique. Le variant étudié ici, appelé L271H, avait été montré dans des cellules non neuronales comme facilitant l’ouverture du canal, mais son impact sur les cellules cérébrales humaines était inconnu.
Construire en laboratoire des cellules cérébrales spécifiques au patient
L’équipe a créé un modèle iPSC dans lequel des cellules sanguines d’une fille porteuse du variant L271H ont été reprogrammées en cellules souches capables de former n’importe quel tissu. Ces cellules souches pluripotentes induites ont ensuite été guidées pour devenir des précurseurs neuronaux — les blocs de construction précoces qui génèrent plus tard des neurones — et des neurones du mésencéphale impliqués dans la signalisation dopaminergique. Les chercheurs ont aussi cultivé des organoïdes corticaux tridimensionnels, de petits « mini‑cerveaux » qui reproduisent les premiers stades de la formation du cortex humain. En comparant les cellules dérivées du patient avec plusieurs lignées témoins provenant de donneurs sains, ils ont pu distinguer les effets du variant de la variation génétique naturelle.
Des cellules précoces trop actives, des neurones matures sous‑actifs
Chez les précurseurs neuronaux les plus précoces, le canal Cav1.3 était la forme dominante de ce type de porte calcique. Les cellules porteuses du variant L271H présentaient des poussées calciques spontanées plus fréquentes que les témoins, malgré des propriétés électriques de base similaires. Cela suggère qu’à ce stade, le variant perturbe principalement l’équilibre calcique intracellulaire plutôt que de provoquer directement des décharges électriques extrêmes. À mesure que les cellules ont mûri en neurones du mésencéphale, le tableau s’est inversé. Bien qu’un autre type de canal calcique soit devenu plus abondant au global, les neurones porteurs du variant L271H manifestaient moins d’événements calciques, un potentiel de repos plus dépolarisé et étaient plus difficiles à amener à déclencher des trains d’activité répétés. Leurs potentiels d’action étaient de plus faible amplitude et plus lents à monter et descendre, indiquant que le variant affaiblit finalement la capacité des neurones à transmettre des signaux électriques robustes.
Des mini‑cerveaux qui croissent plus vite mais s’organisent mal
Dans les organoïdes corticaux, le variant L271H a laissé une empreinte structurelle marquée. Comparés aux témoins, les organoïdes du patient présentaient moins de cavités de type ventricule et ces cavités étaient plus petites — des zones annulaires où les cellules progénitrices s’alignent normalement de manière ordonnée. Les cellules gliales radiales, qui servent d’échafaudage et guident les neurones nouveaux‑nés, étaient disséminées dans le tissu au lieu de former des couches polarisées et régulières. Les progéniteurs intermédiaires et les neurones sont apparus plus tôt puis ont décliné plus rapidement, ce qui suggère que les progéniteurs ont été poussés à se différencier trop vite. Au bout de 60 jours, le nombre total de neurones avait rattrapé le retard, mais le mauvais timing et la désorganisation précoces indiquent un plan de construction perturbé des circuits corticaux plutôt qu’une incapacité absolue à produire des neurones.
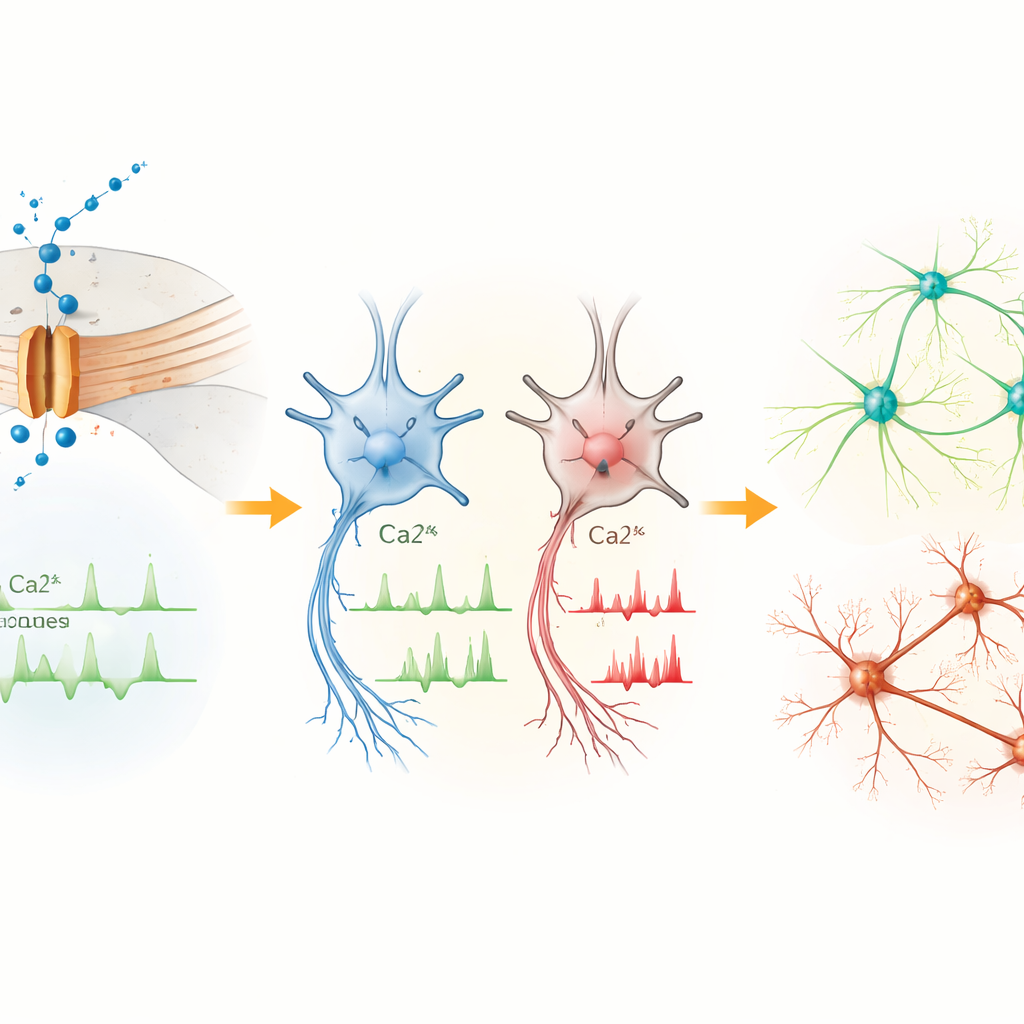
L’activité génétique se réoriente vers des programmes à risque neurodéveloppemental
Pour comprendre comment la gestion altérée du calcium influence le contrôle génétique, l’équipe a séquencé l’ARN des cellules du patient et des témoins à différents stades. Les différences étaient modestes dans les cellules souches mais se sont amplifiées dans les progéniteurs et les neurones à mesure que l’expression de Cav1.3 augmentait. De nombreux gènes régulés à la hausse ou à la baisse dans les cellules du patient participent au contrôle de la transcription, au développement cérébral et à la neurogenèse. Plusieurs d’entre eux, dont PTN, MEIS2, POU3F2, CNTN4, CNTNAP2 et AUTS2, sont des gènes de risque connus pour l’autisme et des conditions neurodéveloppementales apparentées. Ces modifications suggèrent qu’une entrée calcique anormale via Cav1.3 reconfigure des programmes génomiques dépendants de l’activité qui guident la prolifération, la migration et la connectivité des cellules cérébrales.
Ce que cela signifie pour les patients et les thérapies futures
En résumé, ce travail montre comment un seul variant de canal calcique peut d’abord surstimuler les cellules cérébrales précoces, puis laisser les neurones matures sous‑alimentés, tout en décalant des réseaux géniques liés aux troubles du neurodéveloppement. Le résultat est un cerveau qui peut paraître en grande partie normal extérieurement mais dont les circuits se sont développés trop vite et se sont organisés imparfaitement. En recréant la mutation de cette patiente dans des cellules humaines et des organoïdes, l’étude fournit une feuille de route détaillée reliant un défaut moléculaire à une altération du signal cellulaire, de l’architecture tissulaire et de la régulation génique. Ce modèle offre désormais une plateforme pour tester des médicaments ciblés et des stratégies génétiques visant à restaurer une signalisation calcique plus normale dans les canalopathies liées à Cav1.3.
Citation: Tisch, M., Geisler, S.M., Gabassi, E. et al. Aberrant calcium signaling and neuronal activity in the L271H CACNA1D (Cav1.3) iPSC model of neurodevelopmental disease. Mol Psychiatry 31, 2927–2940 (2026). https://doi.org/10.1038/s41380-025-03429-8
Mots-clés: canaux calciques, troubles du neurodéveloppement, cellules souches pluripotentes induites, brain organoids</keyword/organoïdes cérébraux> <keyword>gènes de risque pour l’autisme