Clear Sky Science · pl
PCBP1 reguluje alternatywne składanie AARS2 w wrodzonej kardiomiopatii
Dlaczego niektóre niemowlęta rodzą się z niewydolnymi sercami
U niektórych noworodków ciężka postać niewydolności serca pojawia się krótko po narodzinach, nie z powodu zablokowanych tętnic, lecz z powodu drobnych uchybień w tym, jak komórki serca wytwarzają energię. Lekarze powiązali ten stan z mutacjami w genie AARS2, jednak etapy łączące zmianę w DNA z słabym, nieprawidłowo ukształtowanym sercem pozostawały niejasne. To badanie odkrywa kluczowe brakujące ogniwo: molekularnego „redaktora”, który pomaga komórkom serca prawidłowo składać komunikat AARS2, aby mitochondria — elektrownie komórkowe — mogły utrzymać bijące serca przy życiu.
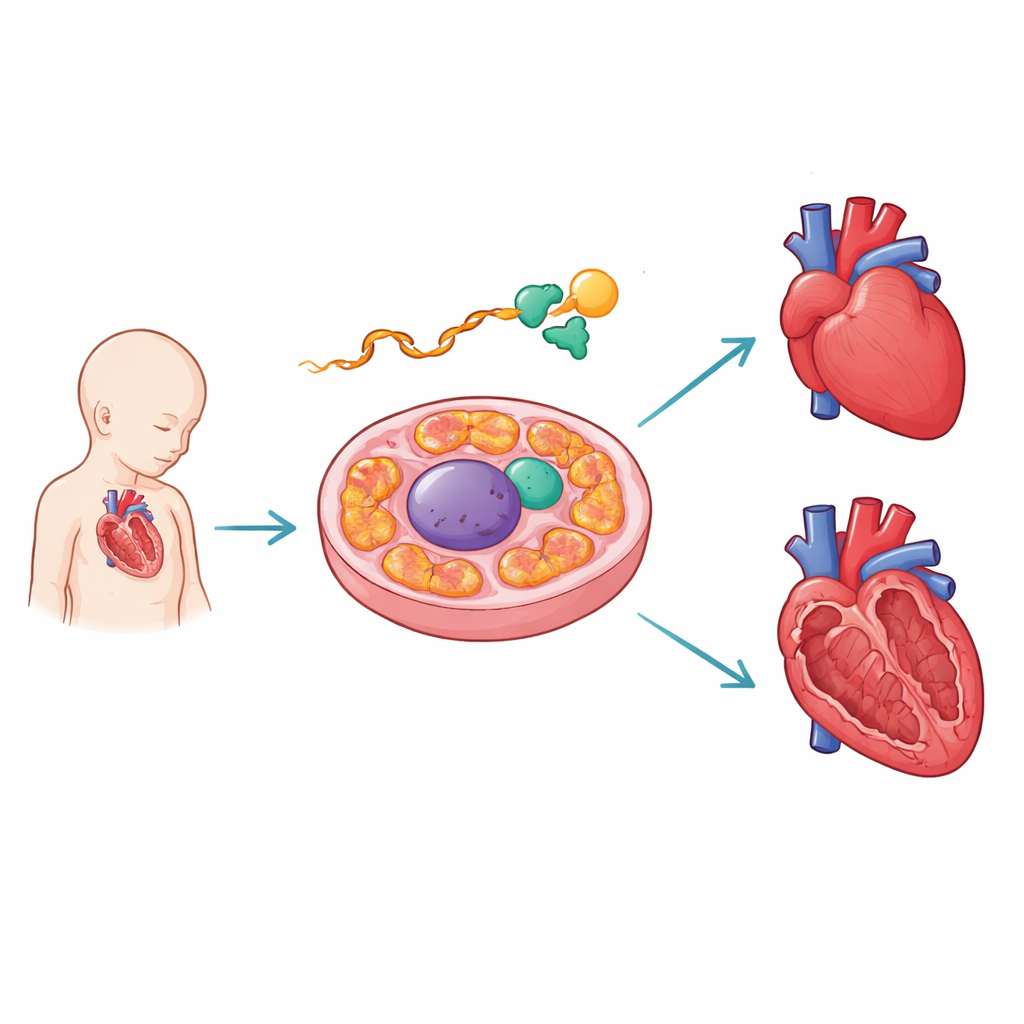
Molekularny redaktor, który chroni rozwijające się serce
Autorzy skupili się na PCBP1, białku wiążącym RNA, pośredniczącym w przekazie informacji skopiowanej z DNA. W rozwijających się sercach myszy stwierdzili, że PCBP1 przyłącza się do RNA AARS2 w pobliżu krytycznego miejsca łączenia. AARS2 pomaga załadować aminokwas alaninę na tRNA w mitochondriach, co jest kluczowym krokiem w budowie białek napędzających produkcję energii. Wykorzystując mapy wiązania o wysokiej przepustowości i konstruując raportery, zespół wykazał, że miejsca kontaktu PCBP1 pokrywają się z ludzkimi mutacjami chorobotwórczymi, które zaburzają dołączanie jednego specyficznego fragmentu — eksonu 16 — komunikatu AARS2. Gdy PCBP1 jest nieobecny lub jego miejsca wiążące są uszkodzone, ten ekson jest pomijany, co niweczy instrukcję tworzenia pełnej długości enzymu AARS2.
Gdy redagowanie zawodzi, struktura serca szwankuje
Aby zobaczyć, co to oznacza dla całego narządu, badacze selektywnie usunęli PCBP1 z komórek mięśnia sercowego myszy. Te serca rozwijały się nieprawidłowo: ich ściany były cienkie i gąbczaste zamiast gęsto zbudowane, a wierzchołek serca często rozdwajał się, tworząc malformację przypominającą u ludzi niekompaktację lewej komory. Wiele myszy zmarło krótko po urodzeniu. Na poziomie RNA setki genów przesunęły się w stronę niemowlęcego, wczesnego embrionalnego wzorca, co wskazywało na opóźnione dojrzewanie jam serca. Co istotne, komunikat Aars2 niemal zawsze pomijał ekson 16, prowadząc do skróconego transkryptu i dramatycznego spadku białka AARS2, co potwierdza, że wadliwy splicing tego jednego genu jest główną konsekwencją utraty PCBP1.
Bezpośrednie uszkodzenie AARS2 odtwarza chorobę
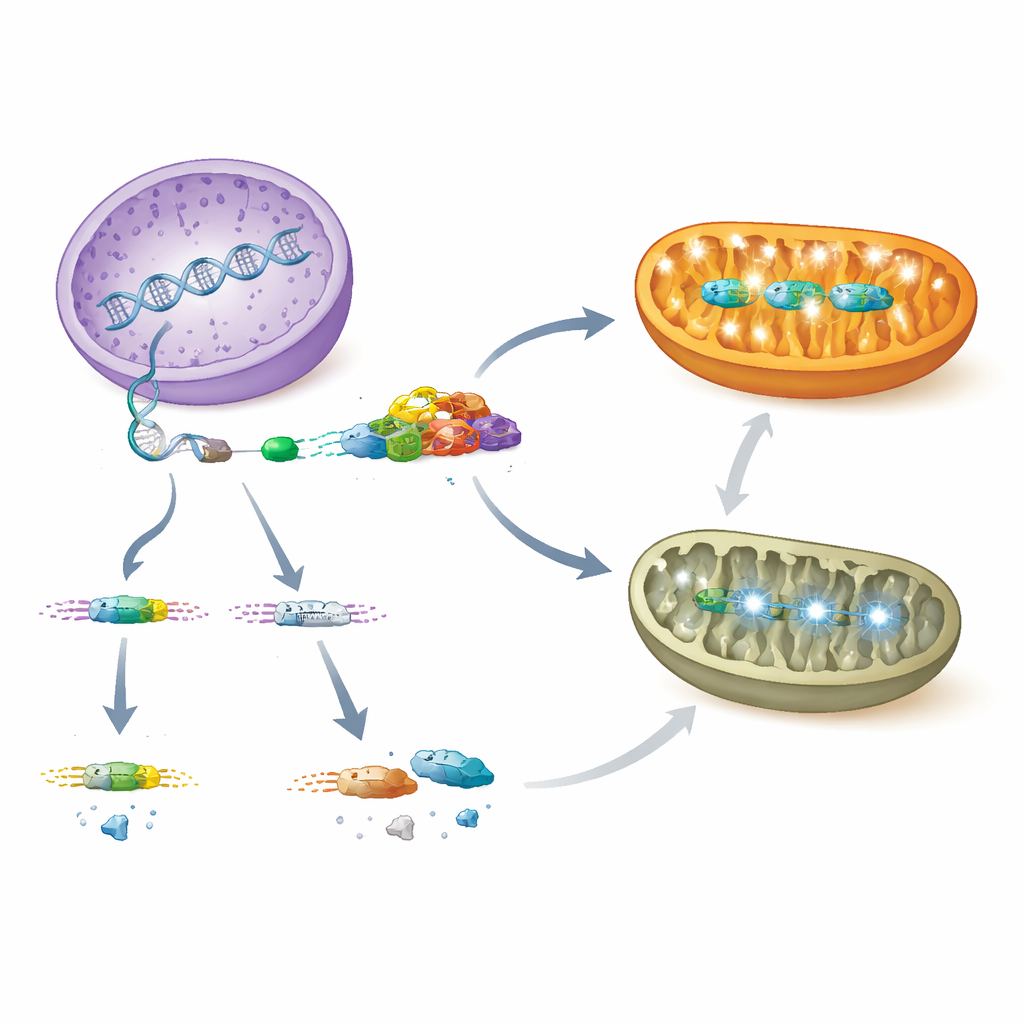
Następnie zespół skonstruował myszy, w których ekson 16 Aars2 mógł zostać usunięty tylko w komórkach mięśnia sercowego. Zwierzęta te bardzo przypominały myszy pozbawione PCBP1: wykazywały zniekształcone ściany komór serca, umierały około narodzin, a ich serca wykazywały ten sam niedojrzały sygnaturowy wzorzec ekspresji genów. Gdy badacze wyzwolili utratę Aars2 później, po urodzeniu, zwierzęta początkowo wydawały się zdrowe, lecz wkrótce rozwinęły powiększone, osłabione serca z rozległymi bliznami. Mikroskopia elektronowa ujawniła, że mitochondria miały mniej wewnętrznych fałdów, a szczegółowe testy biochemiczne pokazały, że kluczowe kompleksy łańcucha oddechowego, szczególnie kompleksy I i IV, zostały zdezorganizowane i straciły aktywność długo przed pojawieniem się jawnej niewydolności serca.
Jak zmagające się mitochondria komunikują się z jądrem komórkowym
Utrata AARS2 sprawiła, że mitochondria komórek serca nie były w stanie utrzymać normalnej fosforylacji oksydacyjnej, głównego sposobu produkcji ATP. Zarówno w sercach pozbawionych PCBP1, jak i w sercach z deficytem Aars2, wiele genów jądrowych kodujących elementy aparatu energetycznego zostało wyciszonych, a odpowiadające im białka mitochondrialne zostały uszczuplone. Jednocześnie komórki włączyły skoordynowany program stresowy znany jako odpowiedź na niepogrupowane białka oraz zintegrowana odpowiedź na stres. Centralne elementy tej ścieżki, w tym białko ATF4 i jego górny regulator, ufosforylowane eIF2α, wzrosły gwałtownie, podobnie jak LONP1, mitochondrialna proteaza usuwająca uszkodzone białka. U starszych mutantów Aars2 badacze zaobserwowali również aktywację regulatora wzrostu MYC i powiększenie jąderka — zakładu produkcji rybosomów — wraz z napływem jądrowo kodowanych czynników translacji mitochondrialnej, co sugeruje, że jądro próbowało zrekompensować upośledzoną syntezę białek mitochondrialnych.
Co to oznacza dla dzieci z rzadką niewydolnością serca
Razem te ustalenia przedstawiają jasną ścieżkę od regulacji genu do choroby narządu. PCBP1 normalnie wiąże się z RNA AARS2 w komórkach serca i zapewnia włączenie eksonu 16, co daje stabilny enzym wspierający syntezę białek mitochondrialnych i silną produkcję energii. Gdy PCBP1 jest utracony lub gdy mutacje zaburzają ten etap splicingu, ekson 16 jest pomijany, poziomy AARS2 się załamują, kompleksy energetyczne mitochondriów rozpadają się, a rozwijające się ściany serca nie ulegają prawidłowej kompaktacji, prowadząc do śmiertelnej kardiomiopatii. Zestresowane mitochondria następnie sygnalizują z powrotem do jądra, uruchamiając programy ochronne, lecz te odpowiedzi nie wystarczają, by zapobiec niewydolności serca. Rozjaśniając oś PCBP1–AARS2, badanie sugeruje nowe markery diagnostyczne i potencjalne cele terapeutyczne dla niemowląt i dzieci z inaczej niewyjaśnioną mitochondrialną chorobą serca.
Cytowanie: Lu, Y.W., Liang, Z., Dorr, K. et al. PCBP1 regulates alternative splicing of AARS2 in congenital cardiomyopathy. Nat Cardiovasc Res 5, 328–350 (2026). https://doi.org/10.1038/s44161-026-00798-3
Słowa kluczowe: mitochondrialna kardiomiopatia, splicing RNA, wrodzona wada serca, gen AARS2, odpowiedź na stres mitochondrialny