Clear Sky Science · de
PCBP1 reguliert alternatives Spleißen von AARS2 bei angeborener Kardiomyopathie
Warum manche Babys mit schwachen Herzen geboren werden
Bei einigen Säuglingen tritt kurz nach der Geburt eine schwere Form von Herzversagen auf, die nicht durch verstopfte Gefäße, sondern durch winzige Fehler in der Energieproduktion ihrer Herzmuskelzellen verursacht wird. Kliniker haben diesen Zustand mit Mutationen im Gen AARS2 in Verbindung gebracht, doch die Schritte, die eine DNA-Veränderung mit einem schwachen, missgebildeten Herzen verknüpfen, blieben lange unklar. Diese Studie deckt eine entscheidende fehlende Verbindung auf: einen molekularen „Editor“, der den AARS2‑Botenstoff in Herzmuskelzellen korrekt zusammenfügt, damit die Mitochondrien – die Kraftwerke der Zelle – das schlagende Herz am Leben erhalten können.
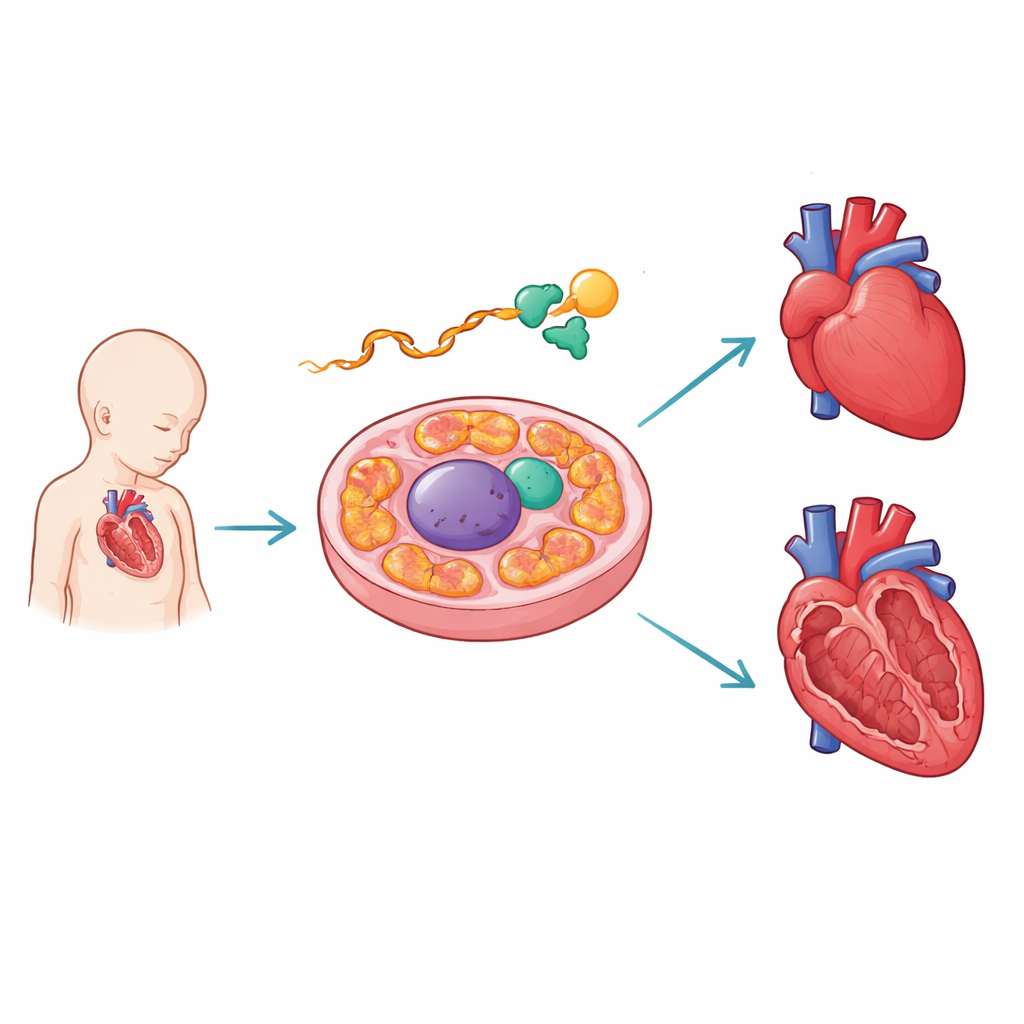
Ein molekularer Editor, der das wachsende Herz schützt
Die Autoren konzentrierten sich auf PCBP1, ein RNA‑bindendes Protein. In sich entwickelnden Mausherzen fanden sie PCBP1 gebunden an die AARS2‑RNA in der Nähe einer kritischen Spleißstelle. AARS2 lädt die Aminosäure Alanin auf Transfer‑RNAs in den Mitochondrien und ist damit ein Schlüsselsschritt beim Aufbau der Proteine, die die Energieproduktion antreiben. Mithilfe hochdurchsatzfähiger Bindungskarten und gentechnisch veränderter Reporterkonstrukte zeigten die Forscher, dass die Bindungsstellen von PCBP1 mit menschlichen Krankheitsmutationen zusammenfallen, die das Zusammenschließen eines bestimmten Abschnitts – Exon 16 – der AARS2‑Botschaft stören. Fehlt PCBP1 oder sind seine Bindungsstellen beschädigt, wird dieses Exon übersprungen und die Anleitung zur Herstellung eines voll funktionsfähigen AARS2‑Enzyms gerät aus der Bahn.
Wenn das Editieren versagt, gerät die Herzstruktur aus der Form
Um zu sehen, was das für ein ganzes Organ bedeutet, entfernten die Forschenden PCBP1 gezielt aus Maus‑Herzmuskelzellen. Diese Herzen entwickelten sich abnorm: Ihre Wände blieben dünn und schwammig statt dicht kompakt, und die Herzspitze teilte sich häufig, eine Fehlbildung, die der menschlichen Erkrankung „linksventrikuläre Non‑Kompaktion“ ähnelt. Viele Mäuse starben kurz nach der Geburt. Auf RNA‑Ebene verschoben Hunderte von Genen ihr Muster in Richtung eines unreifen, frühembryonalen Zustands, was auf eine verzögerte Reifung der Herzkammern hindeutet. Bemerkenswert war, dass die Aars2‑Botschaft fast immer Exon 16 übersprang, was zu einem verkürzten Transkript und einem dramatischen Abfall des AARS2‑Proteins führte und bestätigte, dass fehlerhaftes Spleißen dieses einzelnen Gens eine Hauptfolge des PCBP1‑Verlusts ist.
Direkte Schädigung von AARS2 reproduziert die Krankheit
Anschließend erzeugte das Team Mäuse, bei denen Exon 16 von Aars2 nur in Herzmuskelzellen gelöscht werden konnte. Diese Tiere ahmten die PCBP1‑K.O.-Mäuse nach: Sie zeigten missgebildete Ventrikelwände, starben um die Geburt herum, und ihre Herzen zeigten dasselbe unreife Expressionsmuster. Wenn die Forschenden den Aars2‑Verlust später im Leben mit einem zweiten, postnatalen genetischen Schalter auslösten, wirkten die Tiere zunächst gesund, entwickelten aber bald vergrößerte, geschwächte Herzen mit ausgedehnter Vernarbung. Elektronenmikroskopie zeigte, dass ihre Mitochondrien weniger innere Faltungen hatten, und detaillierte biochemische Tests ergaben, dass Schlüsselkomplexe der Atmungskette, insbesondere Komplex I und IV, destabilisiert waren und ihre Aktivität einbüßten, lange bevor klinisches Herzversagen auftrat.
Wie leidende Mitochondrien mit dem Zellkern kommunizieren
Der Ausfall von AARS2 machte die Mitochondrien der Herzmuskelzellen unfähig, die normale oxidative Phosphorylierung aufrechtzuerhalten, den wichtigsten Weg zur ATP‑Produktion. In sowohl PCBP1‑defizienten als auch Aars2‑defizienten Herzen wurden viele Kern‑kodierte Gene für Komponenten der Energieproduktion herunterreguliert, und die entsprechenden mitochondrialen Proteine waren vermindert. Gleichzeitig schalteten die Zellen ein koordiniertes Stressprogramm ein, bekannt als unförmige Proteinantwort (unfolded protein response) und die integrierte Stressantwort. Zentrale Akteure dieses Pfads, darunter das Protein ATF4 und sein upstream Auslöser, phosphoryliertes eIF2α, stiegen stark an, ebenso wie LONP1, eine mitochondriale Protease, die beschädigte Proteine beseitigt. In älteren Aars2‑Mutanten beobachteten die Forschenden außerdem die Aktivierung des Wachstumsregulators MYC und eine Vergrößerung des Nukleolus – der Fabrik zur Ribosomenproduktion – zusammen mit einem Anstieg kernkodierter mitochondrialer Translationsfaktoren, was darauf hindeutet, dass der Zellkern versucht, den Ausfall der mitochondrialen Proteinproduktion zu kompensieren.
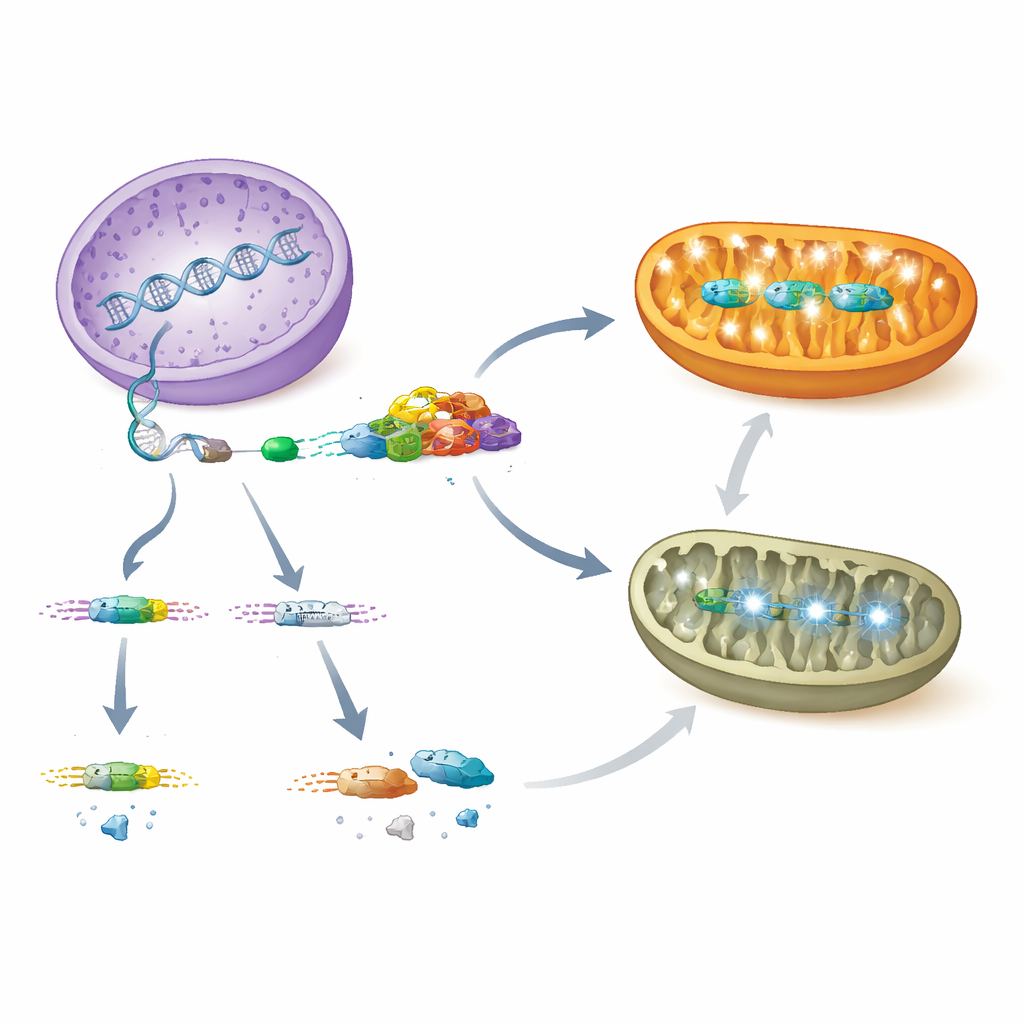
Was das für Kinder mit seltener Herzinsuffizienz bedeutet
Zusammen zeichnen diese Ergebnisse einen klaren Weg von Genregulation zu Organerkrankung nach. PCBP1 bindet normalerweise die AARS2‑RNA in Herzmuskelzellen und stellt sicher, dass Exon 16 inkludiert wird, wodurch ein stabiles Enzym entsteht, das die mitochondriale Proteinsynthese und eine robuste Energieproduktion unterstützt. Geht PCBP1 verloren oder werden durch Mutationen dieser Spleißschritt gestört, wird Exon 16 übersprungen, AARS2‑Spiegel brechen zusammen, mitochondriale Energiekomplexe fallen auseinander und die sich entwickelnden Herzmuskelwände können sich nicht richtig verdichten, was zu tödlicher Kardiomyopathie führt. Die gestressten Mitochondrien senden dann Rückmeldungen an den Zellkern, um Schutzprogramme zu aktivieren, doch diese Reaktionen reichen nicht aus, das Herzversagen zu verhindern. Indem die Studie die PCBP1–AARS2‑Achse beleuchtet, liefert sie neue diagnostische Marker und potenzielle therapeutische Ziele für Säuglinge und Kinder mit sonst rätselhafter mitochondrialer Herzerkrankung.
Zitation: Lu, Y.W., Liang, Z., Dorr, K. et al. PCBP1 regulates alternative splicing of AARS2 in congenital cardiomyopathy. Nat Cardiovasc Res 5, 328–350 (2026). https://doi.org/10.1038/s44161-026-00798-3
Schlüsselwörter: mitochondriale Kardiomyopathie, RNA-Spleißen, angeborener Herzfehler, AARS2-Gen, mitochondriale Stressantwort