Clear Sky Science · it
PCBP1 regola lo splicing alternativo di AARS2 nella cardiomiopatia congenita
Perché alcuni neonati nascono con il cuore che non funziona
Alcuni lattanti sviluppano una forma grave di insufficienza cardiaca subito dopo la nascita, causata non da arterie ostruite ma da piccole anomalie nel modo in cui le cellule cardiache producono energia. I medici hanno collegato questa condizione a mutazioni in un gene chiamato AARS2, ma i passaggi che collegano una variazione del DNA a un cuore debole e malformato sono rimasti oscuri. Questo studio scopre un anello mancante cruciale: un “editor” molecolare che aiuta le cellule cardiache a assemblare correttamente il messaggio di AARS2 affinché i loro mitocondri — le centrali energetiche della cellula — possano mantenere in vita i cuori che battono.
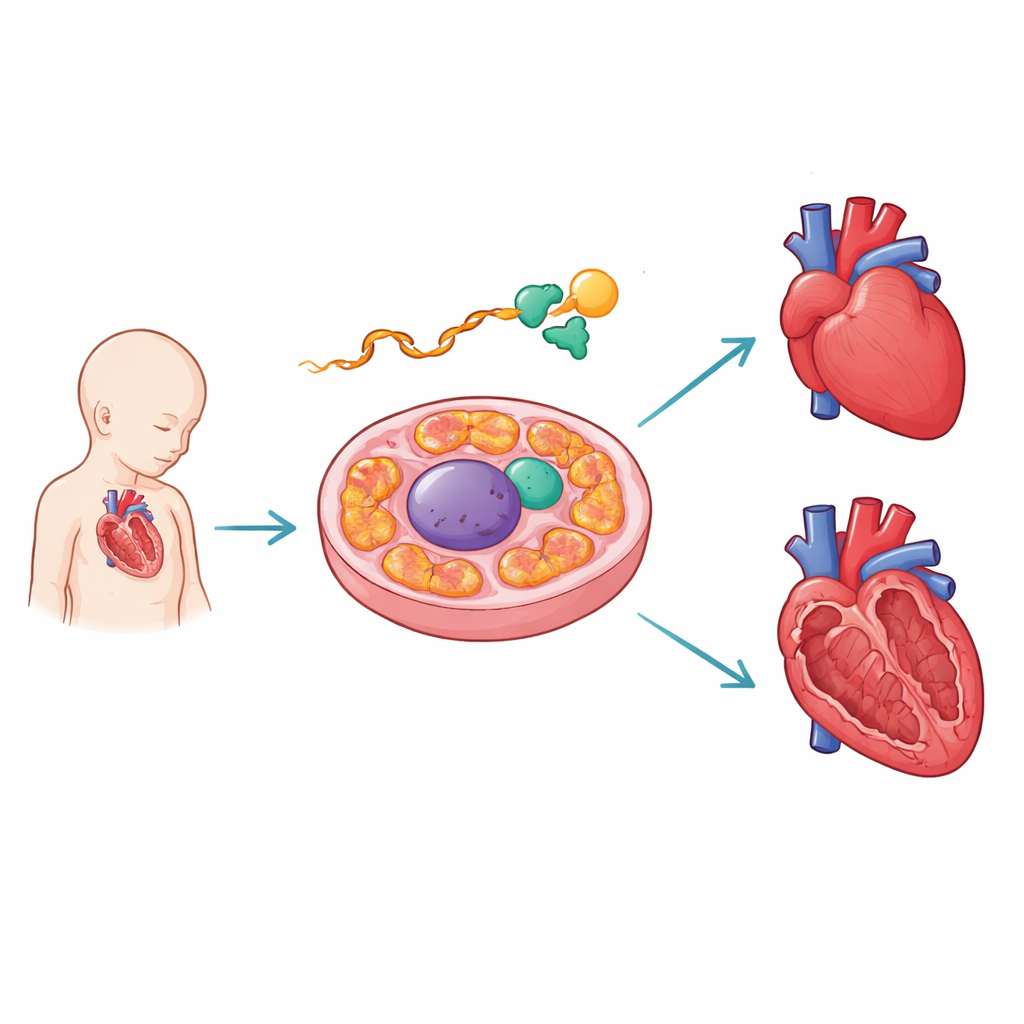
Un editor molecolare che protegge il cuore in crescita
Gli autori si sono concentrati su PCBP1, una proteina che si lega all'RNA, i messaggi intermedi copiati dal DNA. Nei cuori in sviluppo dei topi hanno trovato PCBP1 legarsi all'RNA di AARS2 vicino a una giunzione critica. AARS2 aiuta a caricare l'amminoacido alanina sui trasfer-RNA all'interno dei mitocondri, un passaggio chiave nella costruzione delle proteine che alimentano la produzione di energia. Usando mappe di legame ad alto rendimento e costrutti reporter ingegnerizzati, il gruppo ha dimostrato che i punti di contatto di PCBP1 coincidono con mutazioni umane che interrompono l'unione di un particolare segmento — l'esone 16 — del messaggio di AARS2. Quando PCBP1 manca o i suoi siti di legame sono danneggiati, questo esone viene saltato, compromet tendo le istruzioni per produrre un enzima AARS2 a lunghezza completa.
Quando l'editing fallisce, la struttura cardiaca si altera
Per capire cosa ciò significhi per un intero organo, i ricercatori hanno rimosso selettivamente PCBP1 dalle cellule del muscolo cardiaco nei topi. Questi cuori si sono sviluppati in modo anomalo: le loro pareti erano sottili e spugnose anziché compatte e dense, e la punta del cuore spesso si divideva in due, una malformazione che assomiglia a una condizione umana chiamata non compattazione ventricolare sinistra. Molti topi sono morti poco dopo la nascita. A livello di RNA, centinaia di geni hanno mostrato uno spostamento verso un profilo immaturo, tipico di uno stadio embrionale precoce, indicando un ritardo nella maturazione delle camere cardiache. In particolare, il trascritto di Aars2 quasi sempre saltava l'esone 16, portando a un RNA accorciato e a un calo drammatico della proteina AARS2, confermando che il difetto di splicing di questo singolo gene è una conseguenza principale della perdita di PCBP1.
Danneggiare direttamente AARS2 ricrea la malattia
Successivamente il team ha ingegnerizzato topi in cui l'esone 16 di Aars2 poteva essere eliminato solo nelle cellule del muscolo cardiaco. Questi animali hanno riprodotto fedelmente il fenotipo dei knockout PCBP1: presentavano pareti ventricolari malformate, morivano intorno alla nascita e i loro cuori mostravano lo stesso profilo di espressione genica immaturo. Quando i ricercatori hanno indotto la perdita di Aars2 più tardi nella vita usando un secondo interruttore genetico postnatale, gli animali inizialmente sembravano sani ma hanno presto sviluppato cuori ingrossati e indeboliti con estensive cicatrici. La microscopia elettronica ha rivelato che i loro mitocondri avevano meno pieghe interne, e test biochimici dettagliati hanno mostrato che complessi chiave della catena respiratoria, in particolare i complessi I e IV, erano destabilizzati e avevano perso attività molto prima che comparisse un'insufficienza cardiaca manifesto.
Come i mitocondri in difficoltà comunicano con il nucleo
La perdita di AARS2 ha lasciato i mitocondri delle cellule cardiache incapaci di mantenere una fosforilazione ossidativa normale, il principale modo in cui generano ATP. Sia nei cuori carenti di PCBP1 sia in quelli carenti di Aars2, molti geni nucleari che codificano componenti della macchina energetica sono stati soppressi e le proteine mitocondriali corrispondenti sono risultate impoverite. Allo stesso tempo, le cellule hanno attivato un programma coordinato di stress noto come risposta alle proteine non ripiegate e la risposta integrata allo stress. Attori centrali in questa via, inclusa la proteina ATF4 e il suo innesco a monte, eIF2α fosforilato, sono aumentati sensibilmente, così come LONP1, una proteasi mitocondriale che elimina le proteine danneggiate. Negli Aars2 mutanti più anziani, i ricercatori hanno anche osservato l'attivazione del regolatore della crescita MYC e l'ingrandimento del nucleolo — la fabbrica per la produzione dei ribosomi — insieme a un'impennata nei fattori di traduzione mitocondriale codificati dal nucleo, suggerendo che il nucleo stava cercando di compensare la ridotta produzione di proteine mitocondriali.
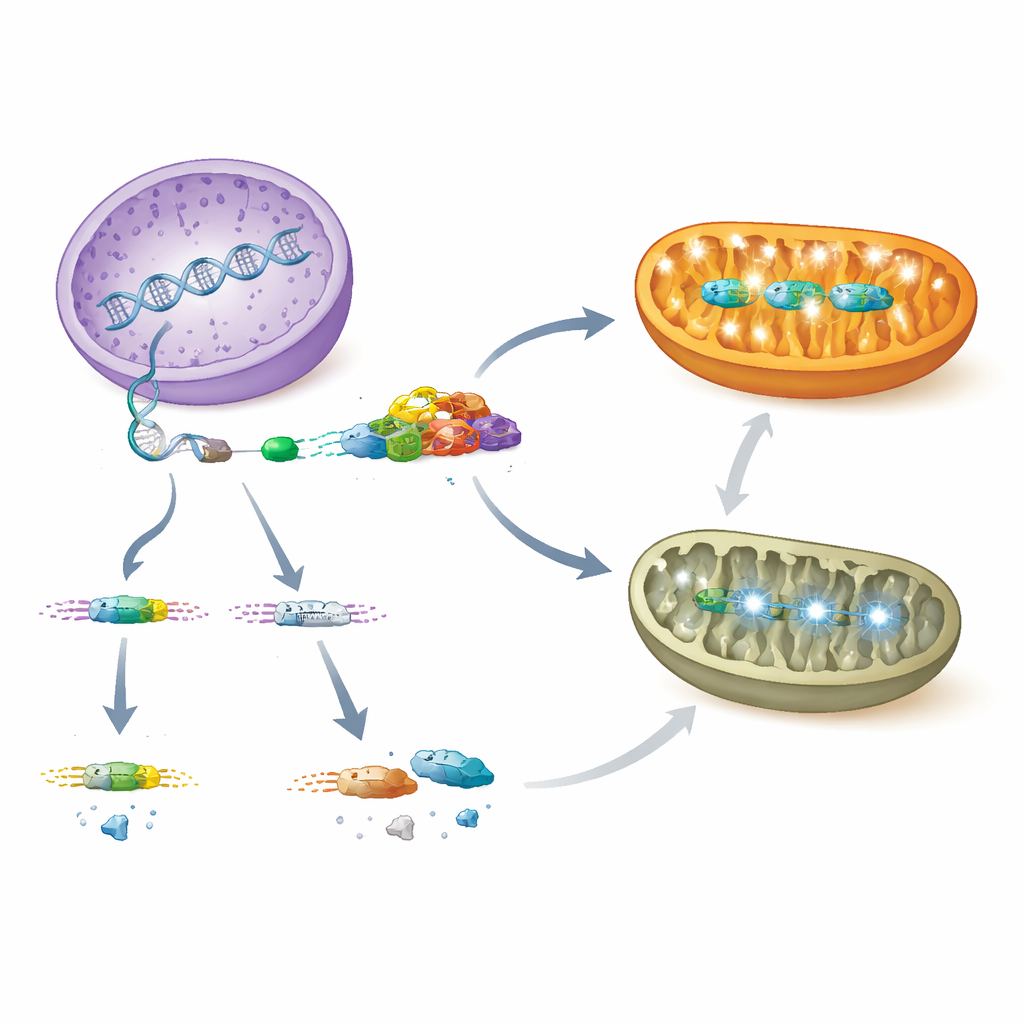
Cosa significa questo per i bambini con insufficienza cardiaca rara
Nel complesso, questi risultati delineano un percorso chiaro dalla regolazione genica alla malattia d'organo. PCBP1 lega normalmente l'RNA di AARS2 nelle cellule cardiache e assicura l'inclusione dell'esone 16, producendo un enzima stabile che sostiene la sintesi proteica mitocondriale e una robusta produzione di energia. Quando PCBP1 viene perso o quando mutazioni interrompono questo passaggio di splicing, l'esone 16 viene saltato, i livelli di AARS2 crollano, i complessi energetici mitocondriali si disfano e le pareti cardiache in sviluppo non si compattano correttamente, portando a una cardiomiopatia letale. I mitocondri in stress segnalano quindi al nucleo per attivare programmi protettivi, ma queste risposte non sono sufficienti a prevenire l'insufficienza cardiaca. Illuminando questo asse PCBP1–AARS2, lo studio suggerisce nuovi marcatori diagnostici e potenziali bersagli terapeutici per neonati e bambini con una forma altrimenti misteriosa di malattia cardiaca mitocondriale.
Citazione: Lu, Y.W., Liang, Z., Dorr, K. et al. PCBP1 regulates alternative splicing of AARS2 in congenital cardiomyopathy. Nat Cardiovasc Res 5, 328–350 (2026). https://doi.org/10.1038/s44161-026-00798-3
Parole chiave: cardiomiopatia mitocondriale, splicing dell'RNA, cardiopatia congenita, gene AARS2, risposta allo stress mitocondriale