Clear Sky Science · pl
Mitachondria jako schronienie dla białek podatnych na agregację utrudniające ich skuteczne niszczenie
Dlaczego zlepki białek mają znaczenie dla zdrowia mózgu
W wielu chorobach mózgu, takich jak choroba Alzheimera, Parkinsona czy Huntingtona, niektóre białka tracą prawidłową strukturę i zlepiają się wewnątrz komórek nerwowych. Te lepkie skupiska mogą zakłócać kluczowe procesy i ostatecznie doprowadzić do śmierci komórek. Badanie stawia pozornie proste pytanie: skoro komórki dysponują potężnymi systemami usuwania odpadów, dlaczego nie radzą sobie skuteczniej z tymi problematycznymi białkami? Odpowiedź prowadzi do nieoczekiwanego miejsca wewnątrz komórki — mitochondriów, często nazywanych elektrowniami komórkowymi.
Komórkowe służby sprzątające i ich ograniczenia
Nasze komórki nieustannie wytwarzają i rozkładają białka. Gdy białka źle się fałdują, jednym z głównych szlaków usuwania jest układ ubikwityna–proteasom, działający jak taśma z oznaczonym śmieciem i niszczarką. Niewłaściwe białka są najpierw oznaczane małymi cząsteczkami (ubikwityną), a następnie wkładane do cylindrycznych maszyn (proteasomów), które rozdrabniają je na nieszkodliwe fragmenty. System ten sprawdza się szczególnie dobrze we wczesnym wychwytywaniu uszkodzonych białek, zanim utworzą duże, nierozpuszczalne grudki. Jednak w chorobach neurodegeneracyjnych grudki i tak się gromadzą, mimo że system niszczenia może pozostać w dużej mierze sprawny. Ta zagadka sugeruje, że problem może leżeć nie tylko w wydajności „niszczarki”, lecz także w tym, czy odpad rzeczywiście do niej trafia.
Odnajdywanie genetycznych przełączników wpływających na rozkład białek
Aby zbadać, co kontroluje los białek skłonnych do zlepiania się, naukowcy stworzyli specjalną linię komórek ludzkich zawierającą fluorescencyjny reporter zaprojektowany tak, by łatwo się źle fałdować, lecz nie być od razu toksyczny. Następnie użyli przesiewu CRISPR–Cas9 obejmującego cały genom, wyłączając po kolei tysiące genów i sortując komórki według ilości nagromadzonego wadliwego reportera. Pozwoliło to precyzyjnie wskazać geny, które albo ułatwiały komórce niszczenie reportera, albo — przeciwnie — sprawiały, że utrzymywał się on dłużej. Jak można było się spodziewać, wiele trafień obejmowało znane elementy machinerii rozkładu białek przy siateczce śródplazmatycznej, błonowym systemie pełniącym funkcję centrum kontroli jakości. Jednak druga, bardziej zaskakująca grupa genów wskazywała na mitochondria i na czynnik translacyjny zwany eIF5A, znany z wpływu na zachowanie mitochondriów.
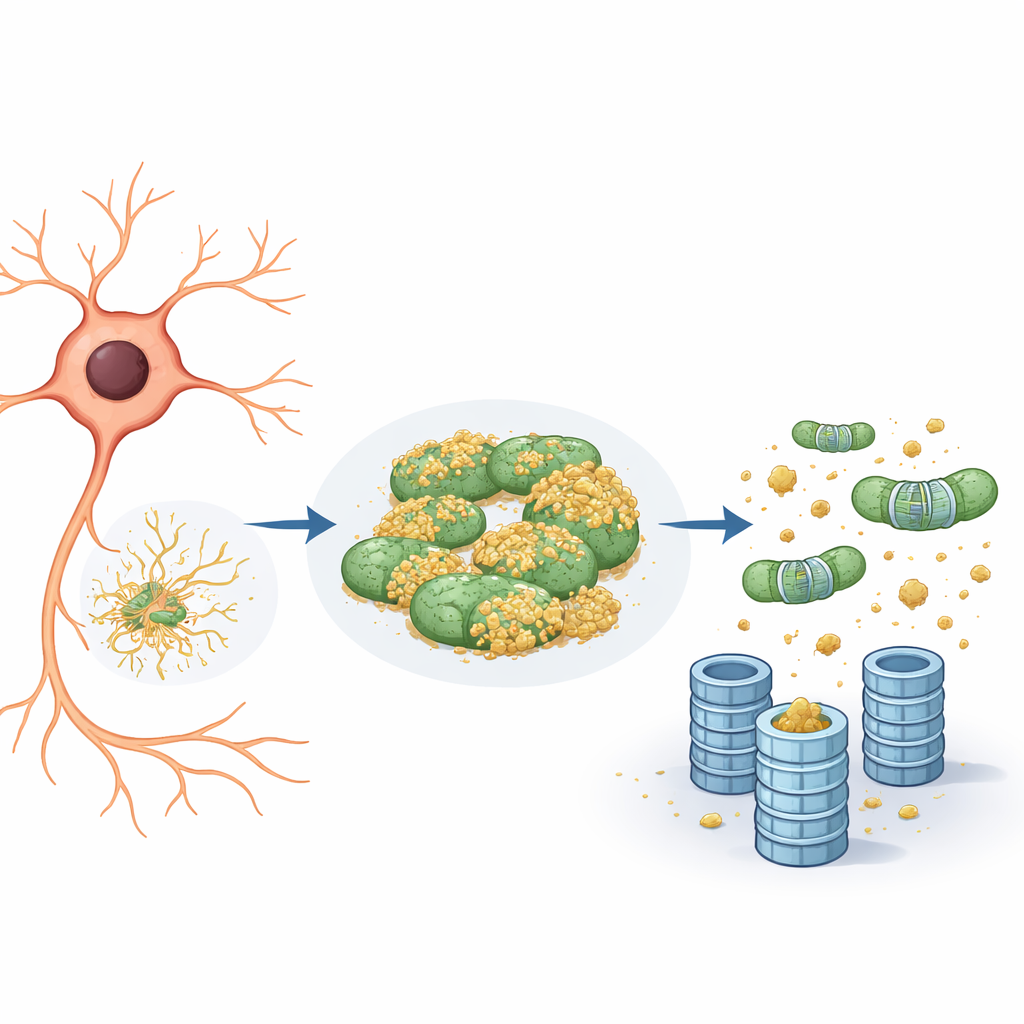
Mitochondria jako kryjówka dla szkodliwych białek
Bardziej szczegółowa analiza wykazała, że reporter białkowy miał tendencję do kojarzenia się nie tylko z siateczką śródplazmatyczną, lecz także z mitochondriami. Na mitochondriach pozostawał w dużej mierze nieoznakowany, a więc niewidoczny dla „niszczarki” proteasomalnej. Gdy zredukowano aktywność eIF5A — albo obniżając jego poziom genetycznie, albo blokując unikalną modyfikację chemiczną niezbędną do jego funkcji — zaszły dwie rzeczy. Po pierwsze, sieć mitochondrialna stała się bardziej pofragmentowana i zreorganizowana. Po drugie, reporter odłączył się od mitochondriów, a jego degradacja przyspieszyła, i to wyłącznie przez układ ubikwityna–proteasom, a nie przez szlaki recyklingu komórkowego, takie jak autofagia. W istocie mitochondria działały jak „schowek”, bezpieczna przystań, w której problematyczne białka mogły uniknąć oznakowania i zniszczenia.
Od sztucznych reporterów do białek powiązanych z chorobami
Klucz do tego zachowania okazał się motyw strukturalny zwany helisą amfipatyczną — krótki fragment częściowo lubiący wodę, a częściowo lipidy, który pomaga białkom przyczepiać się do błon. Gdy badacze „zmiękczyli” tę helisę w swoim reporterze, białko rzadziej wiązało się z mitochondriami lub tworzyło agregaty i przestało wykazywać przyspieszoną degradację po zablokowaniu eIF5A. Co ważne, kilka białek związanych z chorobami, w tym zmutowany huntingtyna (w chorobie Huntingtona) i zmutowany α‑synuklein (w chorobie Parkinsona), także zawiera helisy amfipatyczne i jest znanych z nieprawidłowego lokalizowania się w mitochondriach. W modelach komórkowych ekspresji tych białek, obniżenie aktywności eIF5A lub w inny sposób zaburzenie sieci mitochondrialnej powodowało odłączenie białek chorobowych od mitochondriów i ich bardziej efektywne usuwanie przez proteasomy, przy zmniejszeniu odsetka komórek zawierających duże agregaty.
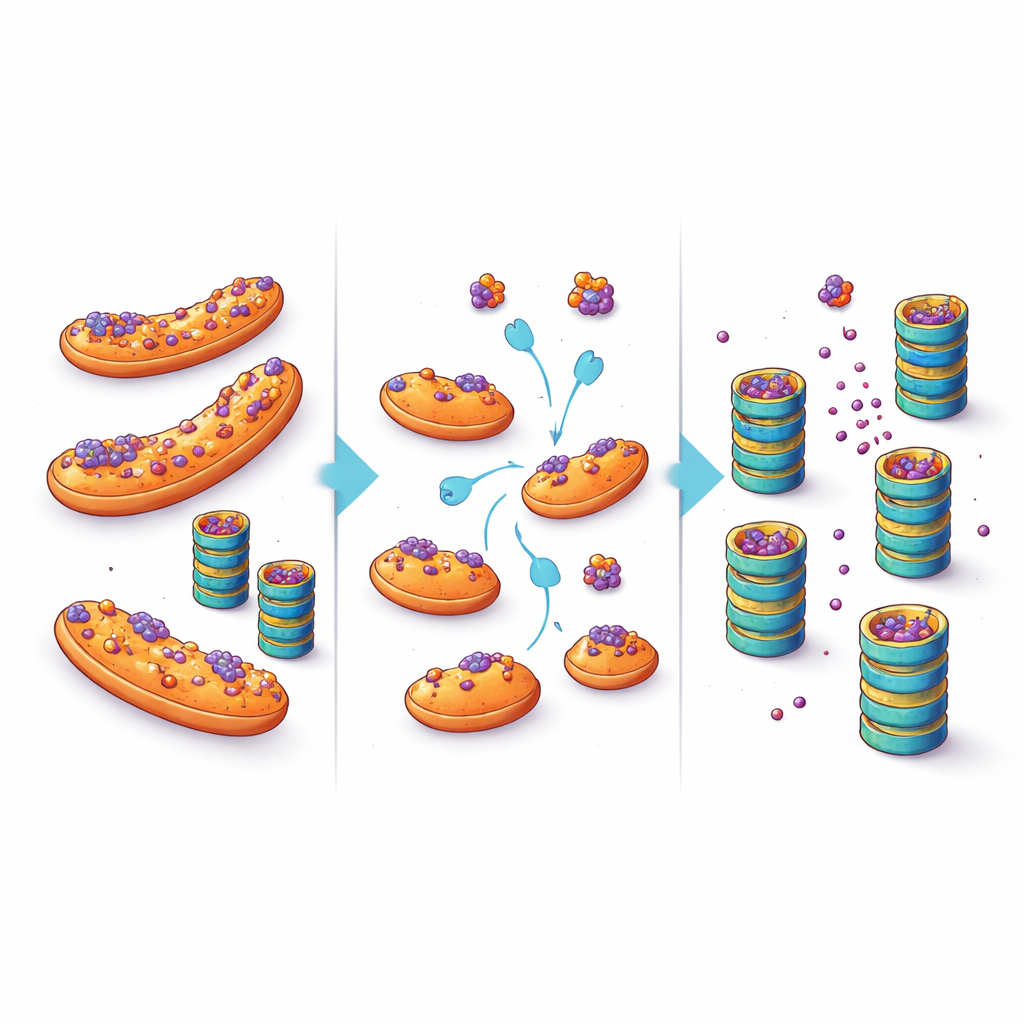
Przekształcenie komórkowej przystani w okazję terapeutyczną
Wyniki te sugerują zmianę paradygmatu myślenia o nagromadzeniu białek w chorobach neurodegeneracyjnych. Zamiast zakładać, że komórkowe „niszczarki” po prostu są przeciążone lub uszkodzone, praca ta podkreśla znaczenie lokalizacji: gdy białka skłonne do agregacji zatrzymują się na mitochondriach, mają utrudniony dostęp do enzymów kontroli jakości i degradują się wolniej. Przesuwając je z tej przystani — poprzez celowane modyfikacje czynników takich jak eIF5A lub bezpośrednie ingerowanie w ich zdolność do dokowania na mitochondriach — może być możliwe obniżenie poziomów toksycznych białek w wrażliwych neuronach. Choć każda przyszła terapia musiałaby zrównoważyć takie interwencje z wieloma innymi rolami mitochondriów i eIF5A, badanie otwiera obiecującą drogę terapeutyczną: zamiast jedynie zwiększać moc komórkowej niszczarki, pomóc źle sfałdowanym białkom dotrzeć do niej.
Cytowanie: Gierisch, M.E., Barchi, E., Marogna, M. et al. Mitochondria serve as a holdout compartment for aggregation-prone proteins hindering efficient degradation. Nat Commun 17, 4195 (2026). https://doi.org/10.1038/s41467-026-72783-0
Słowa kluczowe: mitochondria, agregacja białek, proteasom, neurodegeneracja, eIF5A