Clear Sky Science · it
I mitocondri fungono da compartimento rifugio per proteine inclini all’aggregazione, ostacolando una degradazione efficiente
Perché i grumi proteici sono importanti per la salute del cervello
In molti disturbi cerebrali come Alzheimer, Parkinson e Huntington, alcune proteine perdono la loro conformazione corretta e si aggregano all’interno dei neuroni. Questi aggregati appiccicosi possono disturbare processi vitali e infine uccidere le cellule. Questo studio pone una domanda apparentemente semplice: se le cellule dispongono già di potenti sistemi di smaltimento, perché non eliminano più efficacemente queste proteine problematiche? La risposta conduce a un luogo inaspettato all’interno della cellula: i mitocondri, spesso definiti le centrali energetiche della cellula.
Le squadre di pulizia cellulare e i loro limiti
Le nostre cellule producono e degradano costantemente proteine. Quando le proteine si disfanno, una via principale di smaltimento è il sistema ubiquitina–proteasoma, che funziona come un nastro trasportatore con etichettatura dei rifiuti e trituratore. Le proteine malfunzionanti vengono innanzitutto marcate con piccole molecole (ubiquitina) e poi immessi in macchine a forma di barile (proteasomi) che le scompongono in frammenti innocui. Questo sistema è particolarmente abile nell’intercettare proteine danneggiate precocemente, prima che si induriscano in grandi aggregati insolubili. Eppure, nelle malattie neurodegenerative, gli aggregati si accumulano comunque, anche quando questo sistema di smaltimento può restare in gran parte funzionale. Questo enigma suggerisce che il problema potrebbe non risiedere solo nella forza del trituratore, ma anche nel fatto che la spazzatura raggiunga effettivamente quest’ultimo.
Individuare interruttori genetici che modificano la degradazione proteica
Per esplorare cosa controlla il destino delle proteine inclini all’aggregazione, i ricercatori hanno creato una linea cellulare umana speciale con una proteina reporter fluorescente progettata per disfarsi facilmente, ma senza risultare immediatamente tossica. Hanno quindi eseguito uno screening genomico CRISPR–Cas9, disattivando migliaia di geni uno a uno e ordinando le cellule in base alla quantità del reporter difettoso accumulata. Questo ha permesso di identificare geni che o facilitavano la distruzione del reporter o, al contrario, ne prolungavano la persistenza. Come previsto, molti risultati erano già noti come parti della macchina di smaltimento proteico sulla superficie del reticolo endoplasmatico, un sistema membranoso che funge anche da centro di controllo qualità. Ma un altro gruppo, più sorprendente, puntava verso i mitocondri e verso un fattore di traduzione chiamato eIF5A, noto per influenzare il comportamento mitocondriale.
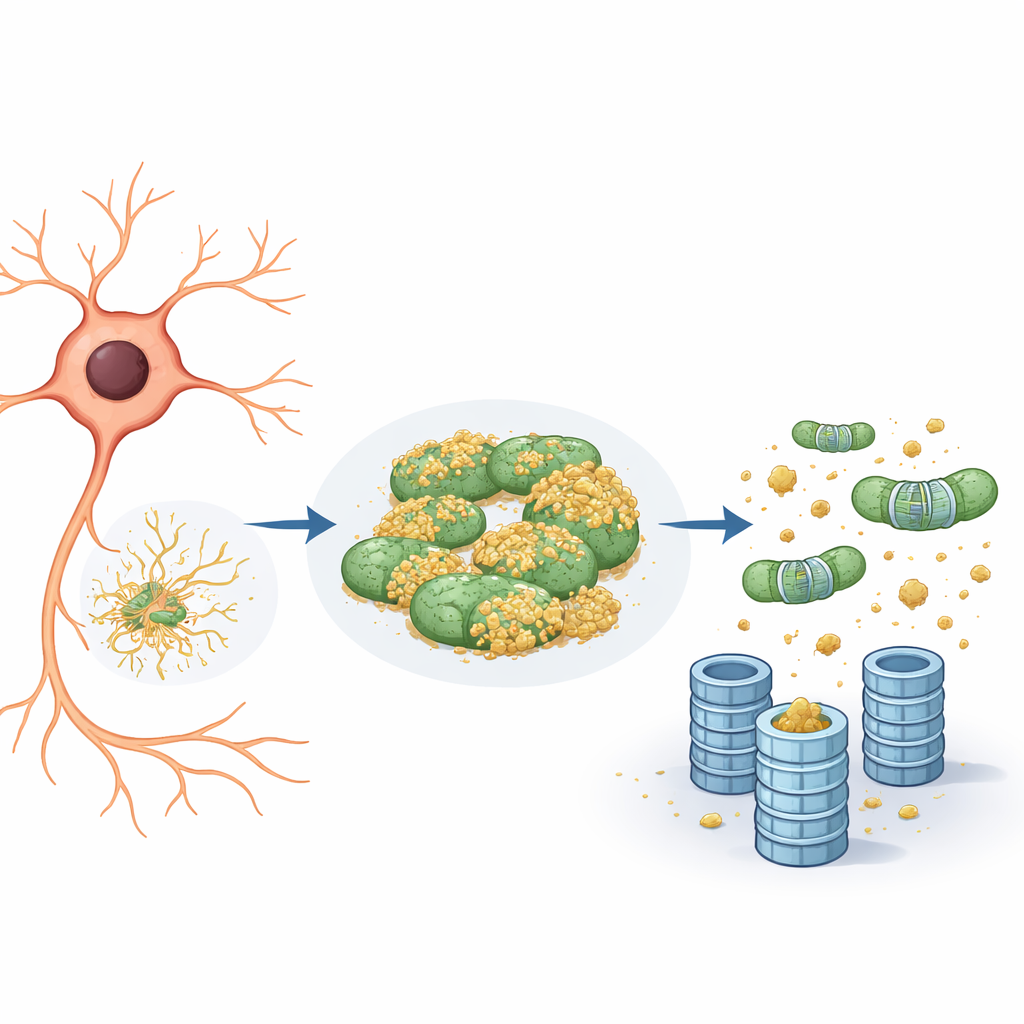
I mitocondri come nascondiglio per proteine difettose
Un’ispezione più ravvicinata ha rivelato che la proteina reporter tendeva ad associarsi non solo al reticolo endoplasmatico ma anche ai mitocondri. Sui mitocondri rimaneva in gran parte non marcata e quindi invisibile al trituratore proteasomico. Quando il team ridusse l’attività di eIF5A—o abbassandone i livelli geneticamente o bloccando una modifica chimica unica necessaria al suo funzionamento—si verificarono due effetti. Primo, la rete mitocondriale divenne più frammentata e si riorganizzò. Secondo, la proteina reporter si staccò dai mitocondri e la sua degradazione accelerò, esclusivamente attraverso il sistema ubiquitina–proteasoma e non tramite vie di riciclo cellulare come l’autofagia. In sostanza, i mitocondri agivano come un “compartimento rifugio”, un porto sicuro dove proteine problematiche potevano evitare di essere marcate e distrutte.
Dai reporter artificiali alle proteine correlate alle malattie
La chiave di questo comportamento si è rivelata essere un motivo strutturale chiamato elica anfipatica—un segmento corto, in parte idrofilo e in parte lipofilo, che aiuta le proteine ad aderire alle membrane. Quando i ricercatori attenuarono questa elica nel loro reporter, la proteina risultò meno propensa a legarsi ai mitocondri o a formare aggregati e non mostrò più un aumento della degradazione quando eIF5A veniva bloccato. È importante che diverse proteine legate alle malattie, inclusa la huntingtina mutata (nella malattia di Huntington) e l’α‑sinucleina mutata (nel morbo di Parkinson), contengano anch’esse eliche anfipatiche e sono note per mislocalizzarsi ai mitocondri. In modelli cellulari che esprimono queste proteine, abbassare l’attività di eIF5A o disturbare la rete mitocondriale fece sì che le proteine patologiche si dissociassero dai mitocondri e venissero eliminate più efficacemente dai proteasomi, con meno cellule che mostravano grandi aggregati.
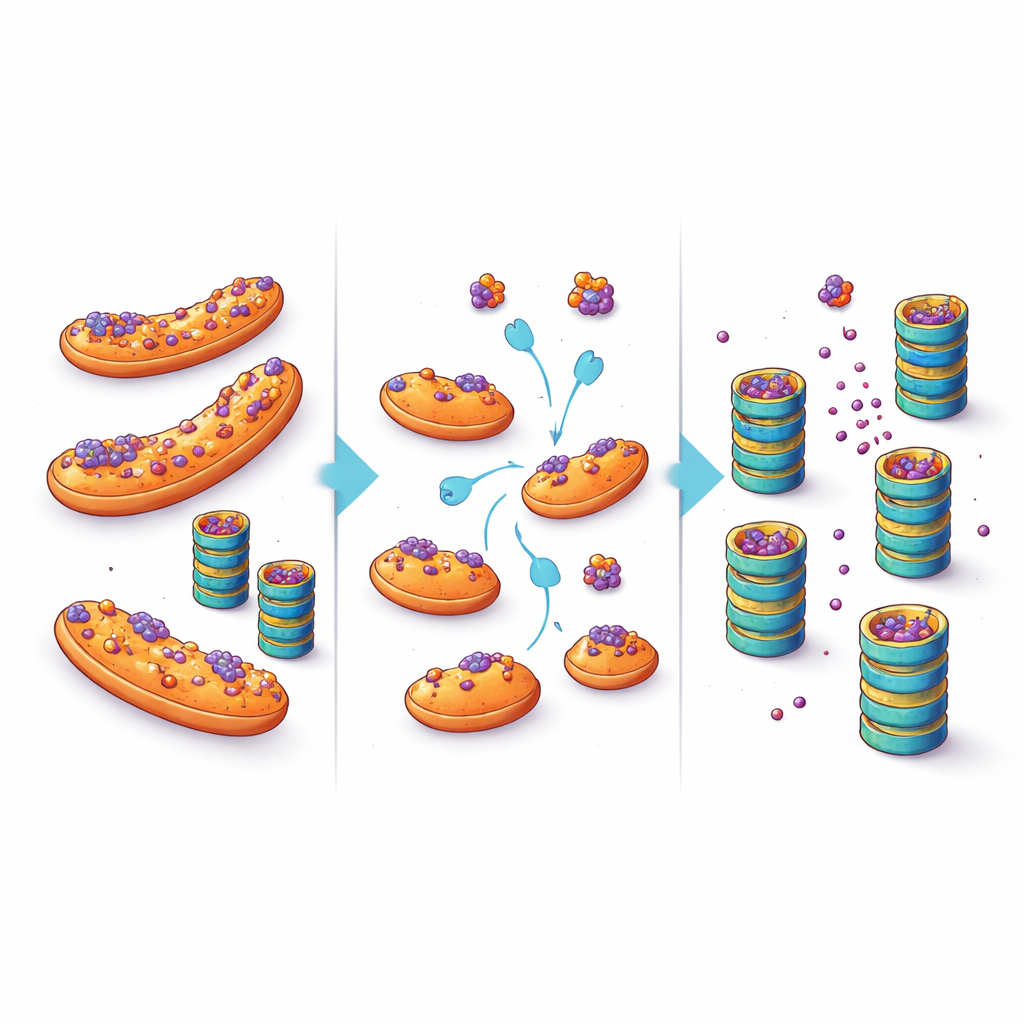
Trasformare un rifugio cellulare in un’opportunità terapeutica
Questi risultati suggeriscono un cambiamento nel modo di pensare all’accumulo proteico nelle malattie neurodegenerative. Piuttosto che assumere che i trituratori cellulari siano semplicemente sovraccarichi o guasti, questo lavoro mette in luce la posizione come fattore cruciale: quando le proteine inclini all’aggregazione sono parcheggiate sui mitocondri, sono meno accessibili agli enzimi di controllo qualità e si degradano più lentamente. Indirizzandole lontano da questo rifugio—attraverso modifiche mirate a fattori come eIF5A o interferendo direttamente con la loro capacità di ancorarsi ai mitocondri—potrebbe essere possibile ridurre i livelli di proteine tossiche nei neuroni vulnerabili. Sebbene qualsiasi trattamento futuro debba bilanciare tali interventi con i numerosi altri ruoli dei mitocondri e di eIF5A, lo studio apre una via terapeutica allettante: invece di limitarsi ad aumentare la potenza del trituratore cellulare, aiutare le proteine malripiegate a raggiungerlo.
Citazione: Gierisch, M.E., Barchi, E., Marogna, M. et al. Mitochondria serve as a holdout compartment for aggregation-prone proteins hindering efficient degradation. Nat Commun 17, 4195 (2026). https://doi.org/10.1038/s41467-026-72783-0
Parole chiave: mitocondri, aggregazione proteica, proteasoma, neurodegenerazione, eIF5A