Clear Sky Science · en
Mitochondria serve as a holdout compartment for aggregation-prone proteins hindering efficient degradation
Why protein clumps matter for brain health
In many brain disorders such as Alzheimer’s, Parkinson’s and Huntington’s disease, certain proteins lose their proper shape and clump together inside nerve cells. These sticky clumps can disrupt vital processes and eventually kill the cells. This study asks a deceptively simple question: if cells already have powerful garbage-disposal systems, why don’t they just get rid of these problem proteins more efficiently? The answer leads to an unexpected place inside the cell—the mitochondria, often called the cell’s power plants.
Cellular cleanup crews and their limits
Our cells constantly make and destroy proteins. When proteins misfold, a major disposal route is the ubiquitin–proteasome system, which works like a tagged-trash and shredder conveyor belt. Misbehaving proteins are first marked with small molecules (ubiquitin) and then fed into barrel-shaped machines (proteasomes) that chew them up into harmless pieces. This system is especially good at catching damaged proteins early, before they harden into large, insoluble clumps. Yet in neurodegenerative diseases, clumps still accumulate, even though this disposal system can remain largely functional. That puzzle suggests that the problem may lie not only in the strength of the shredder, but also in whether the trash actually reaches it.
Finding genetic switches that change protein breakdown
To explore what controls the fate of clump-prone proteins, the researchers created a special human cell line with a fluorescent reporter protein designed to misfold easily, but without being immediately toxic. They then used a genome-wide CRISPR–Cas9 screen, turning off thousands of genes one by one and sorting cells by how much of the faulty reporter they accumulated. This allowed them to pinpoint genes that either helped the cell destroy the reporter or, conversely, made it hang around longer. As expected, many hits were already known parts of the protein-disposal machinery on the surface of the endoplasmic reticulum, a membrane system that doubles as a quality-control hub. But another, more surprising group of genes pointed to the mitochondria and to a translation factor called eIF5A, known to influence mitochondrial behavior.
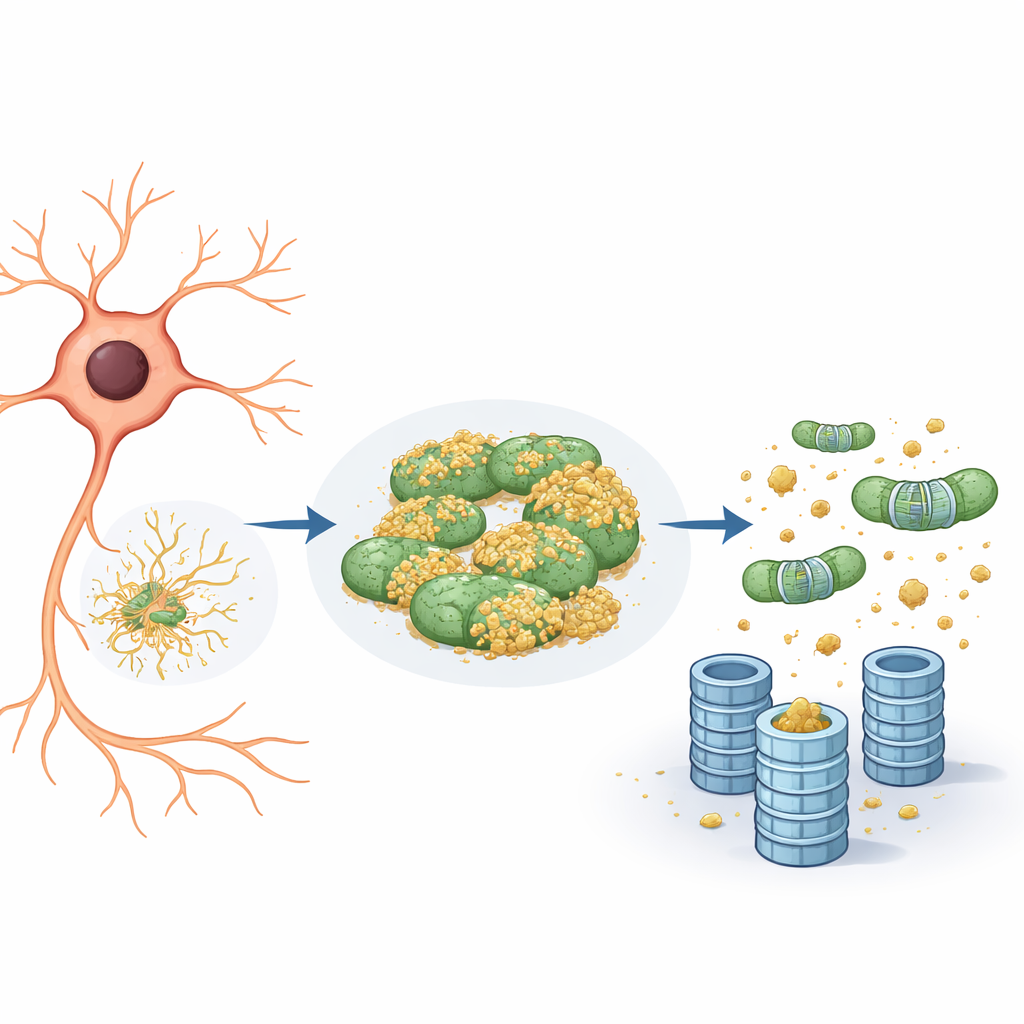
Mitochondria as a hiding place for bad proteins
Closer inspection revealed that the reporter protein tended to associate not only with the endoplasmic reticulum but also with mitochondria. On the mitochondria, it remained largely untagged and thus invisible to the proteasome shredder. When the team reduced the activity of eIF5A—either by lowering its levels genetically or by blocking a unique chemical modification it needs to function—two things happened. First, the mitochondrial network became more fragmented and reorganized. Second, the reporter protein detached from mitochondria and its degradation sped up, strictly through the ubiquitin–proteasome system rather than through cellular recycling pathways like autophagy. In essence, mitochondria were acting as a “holdout compartment,” a safe harbor where troublesome proteins could avoid being tagged and destroyed.
From artificial reporters to disease-linked proteins
The key to this behavior turned out to be a structural motif called an amphipathic helix—a short, partly water-loving and partly fat-loving segment that helps proteins stick to membranes. When the researchers softened this helix in their reporter, the protein was less likely to bind mitochondria or form aggregates and no longer showed enhanced breakdown when eIF5A was blocked. Importantly, several disease-related proteins, including mutant huntingtin (in Huntington’s disease) and mutant α‑synuclein (in Parkinson’s disease), also contain amphipathic helices and are known to mislocalize to mitochondria. In cell models expressing these proteins, lowering eIF5A activity or otherwise disturbing the mitochondrial network caused the disease proteins to dissociate from mitochondria and be cleared more efficiently by proteasomes, with fewer cells showing large aggregates.
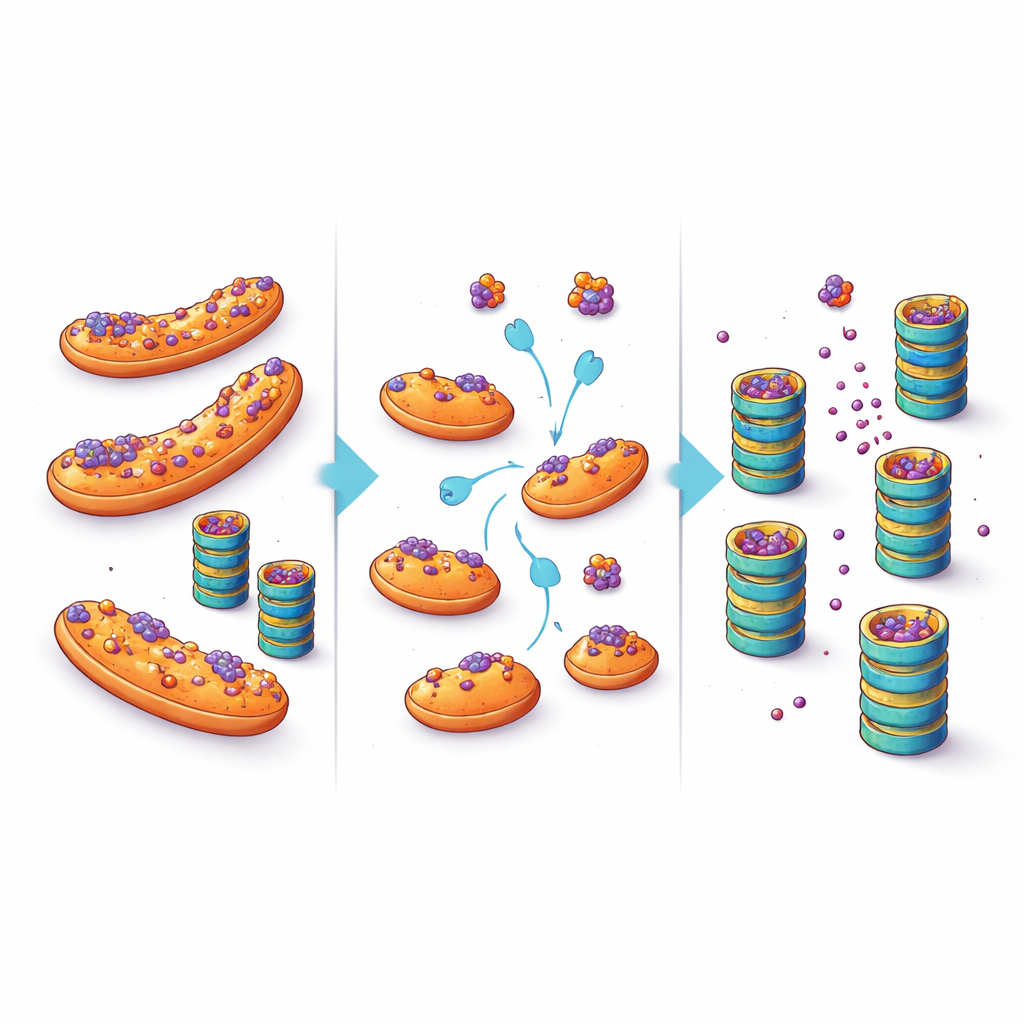
Turning a cellular refuge into a therapeutic opportunity
These findings suggest a shift in how we think about protein buildup in neurodegenerative disease. Rather than assuming the cell’s shredders are simply overwhelmed or broken, this work highlights location as a crucial factor: when aggregation-prone proteins are parked on mitochondria, they are less accessible to quality-control enzymes and degrade more slowly. By nudging them away from this refuge—through targeted changes in factors like eIF5A or by directly interfering with their ability to dock on mitochondria—it may be possible to lower the levels of toxic proteins inside vulnerable neurons. While any future treatment would need to balance such interventions against the many other roles of mitochondria and eIF5A, the study opens an enticing therapeutic avenue: instead of only turning up the power of the cellular shredder, help misfolded proteins find their way into it.
Citation: Gierisch, M.E., Barchi, E., Marogna, M. et al. Mitochondria serve as a holdout compartment for aggregation-prone proteins hindering efficient degradation. Nat Commun 17, 4195 (2026). https://doi.org/10.1038/s41467-026-72783-0
Keywords: mitochondria, protein aggregation, proteasome, neurodegeneration, eIF5A