Clear Sky Science · de
Mitochondrien dienen als Rückzugsort für aggregationsneigende Proteine und behindern deren effiziente Abbau
Warum Proteinklumpen für die Gehirngesundheit wichtig sind
Bei vielen Erkrankungen des Gehirns wie Alzheimer, Parkinson und Huntington verlieren bestimmte Proteine ihre korrekte Faltung und verklumpen innerhalb von Nervenzellen. Diese klebrigen Aggregate können lebenswichtige Prozesse stören und schließlich die Zellen abtöten. Die Studie stellt eine auf den ersten Blick einfache Frage: Wenn Zellen bereits mächtige Müllentsorgungssysteme besitzen, warum beseitigen sie diese problematischen Proteine nicht effizienter? Die Antwort führt an einen unerwarteten Ort in der Zelle — die Mitochondrien, häufig als Kraftwerke der Zelle bezeichnet.
Die zellulären Reinigungstrupps und ihre Grenzen
Unsere Zellen produzieren und bauen ständig Proteine ab. Wenn Proteine fehlgefaltet sind, ist ein wichtiger Entsorgungsweg das Ubiquitin–Proteasom-System, das wie ein markierter Müll und ein Schredderband funktioniert. Fehlverhaltende Proteine werden zuerst mit kleinen Molekülen (Ubiquitin) markiert und dann in fassförmige Maschinen (Proteasomen) gegeben, die sie in harmlose Brocken zerkauen. Dieses System ist besonders gut darin, beschädigte Proteine früh zu erwischen, bevor sie zu großen, unlöslichen Klumpen aushärten. Dennoch akkumulieren in neurodegenerativen Erkrankungen Aggregate, obwohl dieses Entsorgungssystem oft weitgehend intakt bleibt. Dieses Rätsel deutet darauf hin, dass das Problem nicht nur in der Leistungsfähigkeit des Schredders liegt, sondern auch in der Frage, ob der Müll überhaupt zu ihm gelangt.
Genetische Schalter finden, die den Proteinabbau verändern
Um zu untersuchen, was das Schicksal aggregationsgefährdeter Proteine steuert, erzeugten die Forschenden eine spezielle menschliche Zelllinie mit einem fluoreszenten Reporterprotein, das sich leicht fehlfaltet, aber nicht sofort toxisch ist. Anschließend nutzten sie ein genomweites CRISPR–Cas9-Screening, schalteten Tausende Gene einzeln aus und sortierten Zellen danach, wie viel des fehlerhaften Reporters sich anhäufte. Dadurch konnten sie Gene identifizieren, die entweder dem Abbau des Reporters förderlich waren oder umgekehrt dessen Verbleib verlängerten. Wie erwartet waren viele Treffer bereits bekannte Teile der Proteinentsorgungsmaschinerie auf der Oberfläche des endoplasmatischen Retikulums, eines Membransystems, das zugleich als Qualitätskontrollzentrum dient. Eine überraschendere Gruppe von Genen verwies jedoch auf die Mitochondrien und auf einen Translationsfaktor namens eIF5A, der dafür bekannt ist, das mitochondriale Verhalten zu beeinflussen.
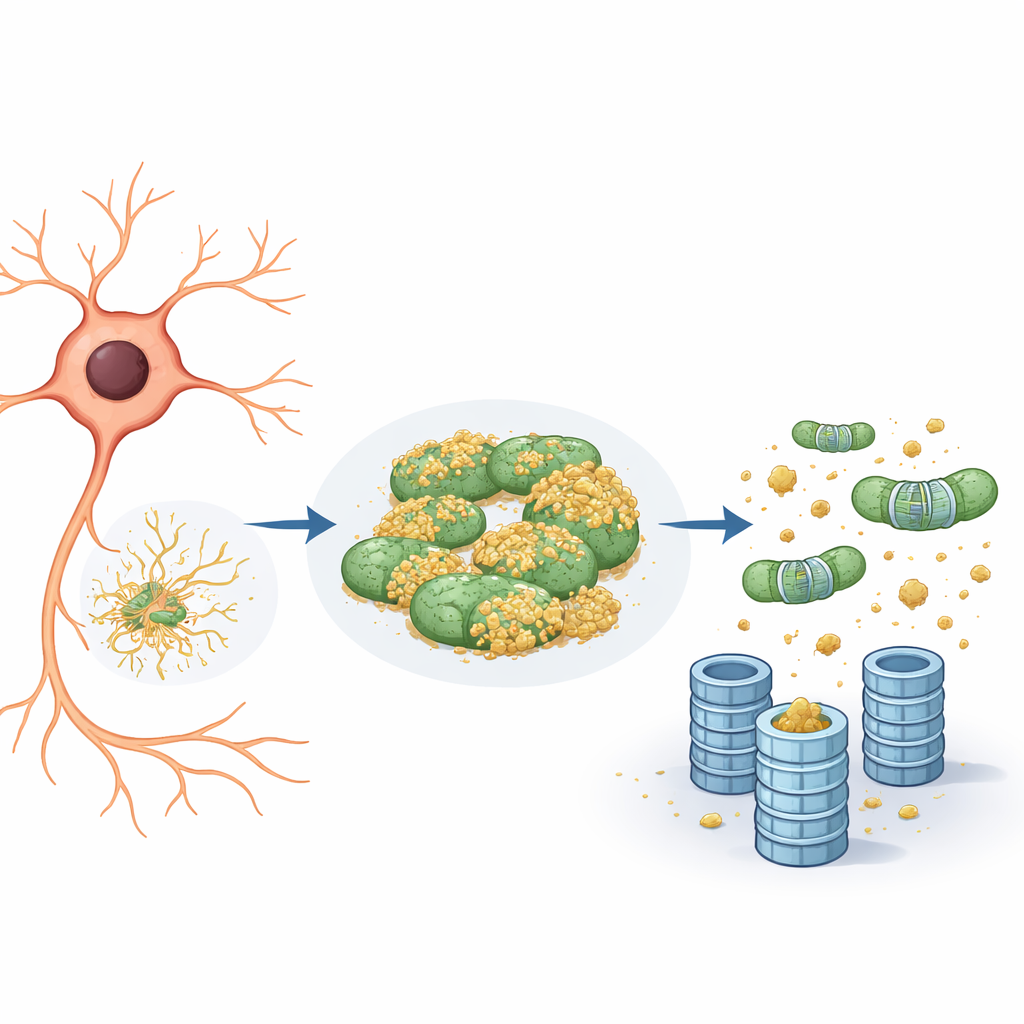
Mitochondrien als Versteck für schädliche Proteine
Genauere Untersuchungen zeigten, dass der Reporter nicht nur mit dem endoplasmatischen Retikulum assoziierte, sondern auch mit Mitochondrien. An den Mitochondrien blieb er größtenteils unmarkiert und war damit für das Proteasom unsichtbar. Reduzierte das Team die Aktivität von eIF5A — entweder durch genetische Verringerung der Menge oder durch Blockade einer für dessen Funktion notwendigen chemischen Modifikation — geschahen zwei Dinge. Erstens wurde das mitochondriale Netzwerk fragmentierter und umorganisiert. Zweitens löste sich der Reporter von den Mitochondrien und sein Abbau beschleunigte sich, strikt über das Ubiquitin–Proteasom-System und nicht über Recyclingwege wie die Autophagie. Im Wesentlichen fungierten Mitochondrien als ein „Rückzugsort“, ein sicherer Hafen, in dem problematische Proteine dem Markieren und Zerstören entgehen konnten.
Von künstlichen Reportern zu krankheitsbezogenen Proteinen
Der Schlüssel zu diesem Verhalten erwies sich als ein Strukturmotiv, die amphipathische Helix — ein kurzes, teils wasserliebendes, teils fettliebendes Segment, das Proteinen hilft, an Membranen zu haften. Wenn die Forschenden diese Helix in ihrem Reporter abschwächten, zeigte das Protein eine geringere Neigung, an Mitochondrien zu binden oder Aggregate zu bilden, und wies auch nicht mehr die verstärkte Abbaurate auf, die bei Blockade von eIF5A beobachtet wurde. Wichtig ist, dass mehrere krankheitsrelevante Proteine, darunter mutiertes Huntingtin (bei Huntington) und mutiertes α‑Synuclein (bei Parkinson), ebenfalls amphipathische Helices enthalten und dafür bekannt sind, sich fehlzuorten und an Mitochondrien zu gehen. In Zellmodellen, die diese Proteine exprimieren, führte eine Verringerung der eIF5A-Aktivität oder anderweitige Störung des mitochondrialen Netzwerks dazu, dass sich die Krankheitsproteine von den Mitochondrien lösten und effizienter durch Proteasomen abgebaut wurden, wobei weniger Zellen große Aggregate zeigten.
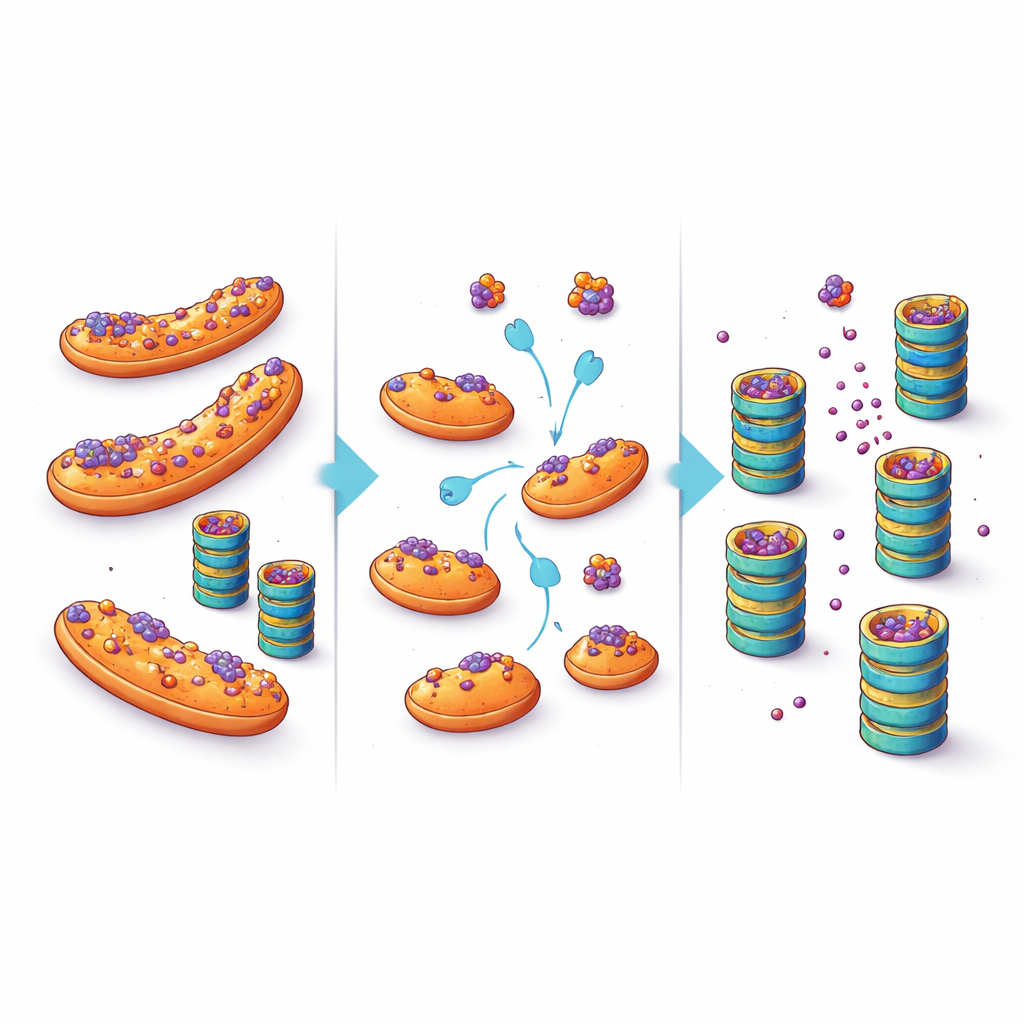
Aus einem zellulären Zufluchtsort eine therapeutische Chance machen
Diese Befunde schlagen eine Verschiebung in der Betrachtung der Proteinansammlung bei neurodegenerativen Erkrankungen vor. Anstatt anzunehmen, die zellulären Schredder seien einfach überfordert oder defekt, hebt diese Arbeit den Standort als entscheidenden Faktor hervor: Wenn aggregationsanfällige Proteine an Mitochondrien geparkt sind, sind sie für Qualitätskontroll-Enzyme schlechter zugänglich und werden langsamer abgebaut. Indem man sie von diesem Zufluchtsort weglenkt — etwa durch gezielte Veränderungen von Faktoren wie eIF5A oder durch direkte Beeinträchtigung ihrer Fähigkeit, an Mitochondrien anzudocken — könnte man die Menge toxischer Proteine in anfälligen Neuronen verringern. Während jede mögliche zukünftige Therapie solche Eingriffe gegen die vielen anderen Rollen von Mitochondrien und eIF5A abwägen müsste, eröffnet die Studie eine verlockende therapeutische Richtung: Statt nur die Kraft des zellulären Schredders zu erhöhen, sollte man fehlgefalteten Proteinen dabei helfen, überhaupt erst dorthin zu gelangen.
Zitation: Gierisch, M.E., Barchi, E., Marogna, M. et al. Mitochondria serve as a holdout compartment for aggregation-prone proteins hindering efficient degradation. Nat Commun 17, 4195 (2026). https://doi.org/10.1038/s41467-026-72783-0
Schlüsselwörter: Mitochondrien, Proteinaggregation, Proteasom, Neurodegeneration, eIF5A