Clear Sky Science · pl
Zablokowanie autofagii powoduje poważne zaburzenia mięśni szkieletowych w modelu myszy miopatii miofibrylarnej 6
Kiedy komórkowy zespół sprzątający zawodzi
Nasze mięśnie nieustannie się naprawiają, w milczeniu usuwając zużyte części, dzięki czemu możemy chodzić, oddychać i poruszać się bez zastanowienia. W artykule omówiono, co się dzieje, gdy ten system porządkowania komórek załamuje się wskutek pojedynczej dziedzicznej mutacji. Skutkiem jest wyniszczająca choroba mięśni u dzieci, osłabiająca kończyny i mięśnie oddechowe. Dzięki szczegółowemu modelowi myszy badacze nie tylko ujawniają, jak uszkodzenia rozwijają się wewnątrz włókien mięśniowych, lecz także testują sposoby ich odwrócenia, dając promyk nadziei na przyszłe terapie.
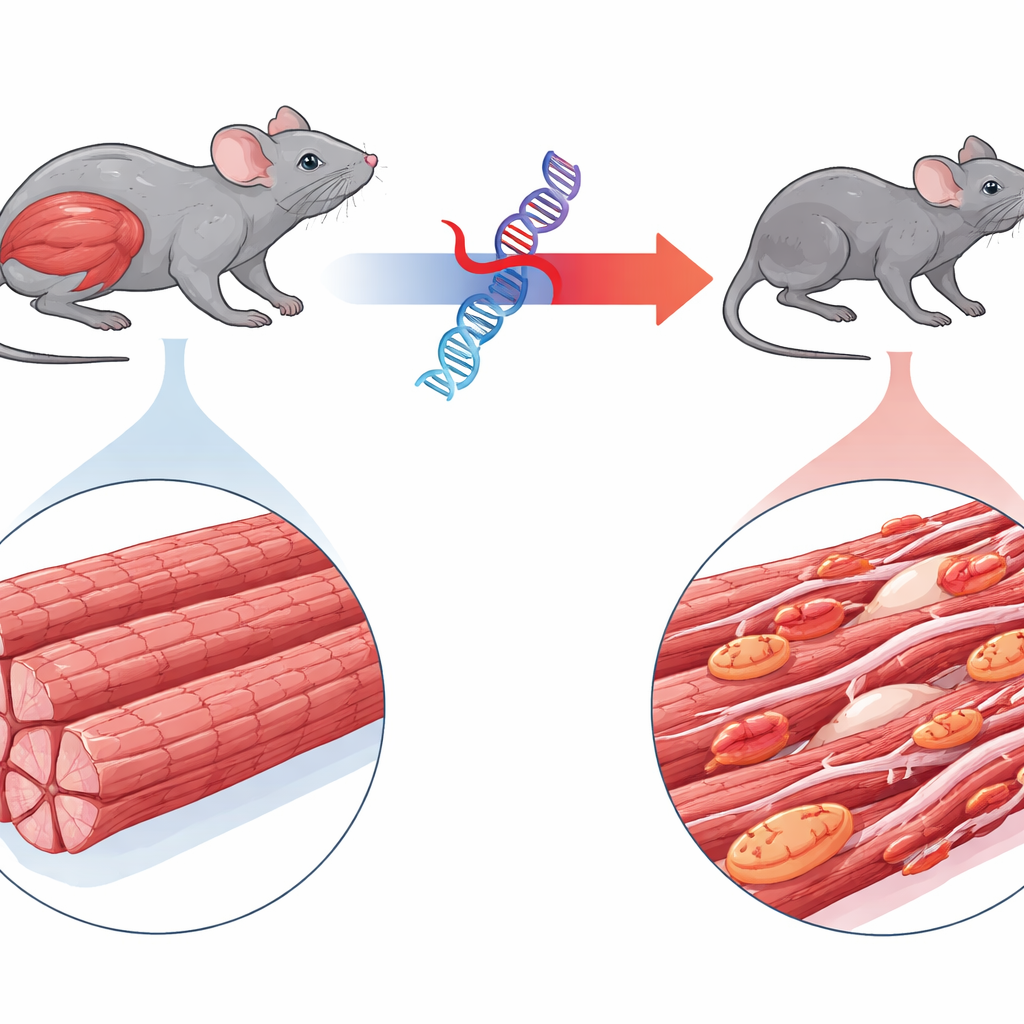
Rzadka choroba mięśni w centrum uwagi
Badanie koncentruje się na miopatii miofibrylarnej 6, rzadkim schorzeniu wywołanym mutacją w genie o nazwie BAG3. Dzieci z tym stanem szybko rozwijają głębokie osłabienie mięśni, czasem problemy z sercem i często umierają w młodym wieku z powodu niewydolności oddechowej. BAG3 zwykle nadzoruje jakość maleńkich jednostek kurczliwych we włóknach mięśniowych, kierując uszkodzone białka do komórkowego recyklingu. Autorzy stworzyli myszy produkujące ludzką, zmutowaną postać BAG3 z przyłączonym fluorescencyjnym markerem, co pozwoliło im śledzić jej zachowanie. W przeciwieństwie do myszy wyrażających normalne ludzkie BAG3, zwierzęta z wersją mutanta szybko stawały się mniejsze, wykazywały zanik mięśni kończyn i oddechowych oraz chód kołyszący się, co ściśle odzwierciedla chorobę u ludzi.
Jak rozpadają się włókna mięśniowe
Badania mikroskopowe i ultrastrukturalne ujawniły, jak gruntownie zmutowane białko zakłóca strukturę mięśnia. U dotkniętych myszy włókna mięśniowe były cieńsze i pełne nieprawidłowych skupisk białek. Wysoce uporządkowane prążki aparatu kurczliwego — sarkomery — były pęknięte i rozmyte, a wiele włókien miało jądra przesunięte do środka, co jest cechą próby regeneracji. Komórki układu odpornościowego naciekały tkankę, a ilość włóknistego materiału bliznowatego wzrosła, choć mniej dramatycznie niż w sercu. Funkcjonalnie, izolowane mięśnie tych myszy wytwarzały do 90 procent mniej siły niż mięśnie zdrowych zwierząt, nawet po uwzględnieniu ich mniejszego rozmiaru, co potwierdza, że podstawowy mechanizm skurczu zawodził.
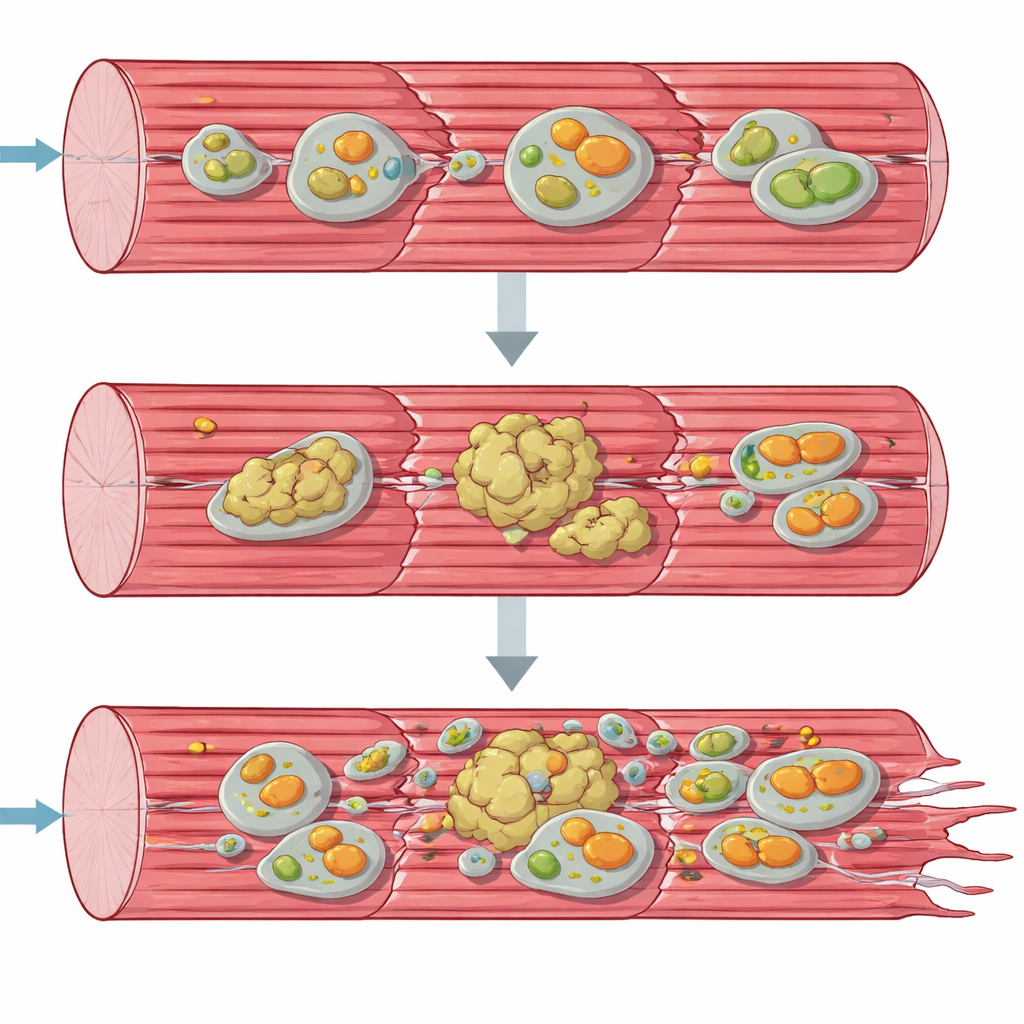
Zatory białkowe i chore mitochondria
Aby zrozumieć rozpad z perspektywy systemowej, zespół sięgnął po narzędzia „omics”, które jednocześnie badają tysiące cząsteczek. Aktywność genów i profile białkowe wykazały, że komórki mięśniowe gorączkowo próbowały reagować: geny odpowiadające za syntezę nowych białek i reakcje na stres były silnie pobudzone, a liczne chaperony i inne czynniki kontroli jakości gromadziły się. Mimo to wiele kluczowych białek strukturalnych aparatu kurczliwego zostało pozbawionych swojej normalnej rozpuszczalnej formy i zamiast tego przesunęło się do frakcji nierozpuszczalnych, bogatych w agregaty. Równocześnie elementy ścieżek recyklingu komórkowego — autofagii i specjalizowanego usuwania uszkodzonych mitochondriów, mitofagii — gromadziły się, jakby utkwiły w połowie procesu. Mikroskopia elektronowa i testy funkcji mitochondriów potwierdziły, że organelle produkujące energię miały zdeformowany kształt, tworzyły skupiska i funkcjonowały znacznie poniżej normalnej wydajności, szczególnie w pierwszym etapie łańcucha oddechowego.
Autofagia jako główny winowajca
Następnie autorzy zapytali, czy podstawowy problem leży w ogólnym komórkowym recyklingu (autofagii), w recyklingu specyficznym dla mitochondriów (mitofagii), czy w obu. Hodowali komórki mięśniowe z zmutowanych myszy i eksperymentalnie wzmacniali różne ścieżki. Pobudzenie szerokiej autofagii białkiem BECN1 usunęło kluczowe markery stresu, zmniejszyło agregaty zmutowanego BAG3 i poprawiło wygląd oraz organizację sarkomerów. W przeciwieństwie do tego wymuszenie mitofagii lub jedynie zwiększenie biogenezy mitochondriów nie skorygowało defektów. U żywych myszy leczenie lekiem aktywującym autofagię — rapamycyną — poprawiło siłę chwytu, koordynację i ruch oraz zwiększyło markery aktywnego recyklingu w mięśniach. Razem te eksperymenty wskazują na pierwotne zablokowanie autofagii, przy czym uszkodzenie mitochondriów pojawia się jako skutek wtórny, a nie źródłowa przyczyna.
Testowanie strategii terapii genowej
Ponieważ choroba jest napędzana przez toksyczne białko, badacze sprawdzili, czy stłumienie zmutowanego BAG3 mogłoby uratować mięśnie. Zapakowali krótkie RNA w kształcie spinki (short hairpin RNA) zaprojektowane tak, by celować specyficznie w ludzki BAG3, do zmodyfikowanego wirusa i podali go do krwiobiegu młodych zmutowanych myszy. Po kilku tygodniach w mięśniach stwierdzono znacznie mniej zmutowanego BAG3, mniej agregatów białkowych, zmniejszone bliznowacenie i znacznie mniej włókien z centralnie położonymi jądrami. Co najważniejsze, leczone mięśnie wytwarzały dramatycznie więcej siły i częściowo odzyskały normalny rozmiar. Ten dowód koncepcji sugeruje, że wyciszanie genu powodującego chorobę, lub połączenie takiego podejścia z lekami pobudzającymi autofagię, mogłoby być skuteczną strategią dla pacjentów, gdy tylko terapia allelo-selektywna zostanie zoptymalizowana.
Co to oznacza dla pacjentów i nie tylko
Prosto ujmując, praca ta pokazuje, że gdy kluczowe białko pomocnicze takie jak BAG3 ulega nieprawidłowemu złożeniu i tworzy grudki, system usuwania odpadów komórkowych zaciąga się. Uszkodzone elementy i wadliwe mitochondria się kumulują, wewnętrzne rusztowanie mięśnia zapada się, a siła wygasa. Dzięki starannemu odtworzeniu choroby u myszy badanie ujawnia, że przywrócenie lub ominięcie tego systemu sprzątającego — albo przez wzmocnienie autofagii, albo przez zmniejszenie ilości zmutowanego BAG3 — może znacząco przywrócić zdrowie mięśni. Choć wiele pozostaje do zrobienia, zanim takie terapie trafią do kliniki, wyniki dostarczają szczegółowej mapy drogowej do zwalczania tej rzadkiej, lecz śmiertelnej choroby mięśni i oferują szersze wnioski o tym, jak zaburzenia komórkowego porządku mogą leżeć u podstaw innych chorób degeneracyjnych.
Cytowanie: Filippi, K., Graf-Riesen, K., Kuppusamy, M. et al. Blockage of autophagy causes severe skeletal muscle disruption in a mouse model for myofibrillar myopathy 6. Nat Commun 17, 3436 (2026). https://doi.org/10.1038/s41467-026-71749-6
Słowa kluczowe: miopatia miofibrylarna, mutacja BAG3, autofagia, degeneracja mięśni szkieletowych, terapia genowa