Clear Sky Science · en
Blockage of autophagy causes severe skeletal muscle disruption in a mouse model for myofibrillar myopathy 6
When the Cell’s Cleanup Crew Fails
Our muscles are constantly repairing themselves, silently clearing away worn-out parts so we can walk, breathe, and move without thinking. This article explores what happens when that cellular cleanup system breaks down because of a single inherited mutation. The result is a devastating childhood muscle disease that weakens limbs and breathing muscles. By building a detailed mouse model, the researchers not only uncover how the damage unfolds inside muscle cells, but also test ways to reverse it, offering a glimmer of hope for future therapies.
A Rare Muscle Disease in Focus
The study centers on myofibrillar myopathy 6, a rare disorder caused by a mutation in a gene called BAG3. Children with this condition quickly develop profound muscle weakness, sometimes heart problems, and often die young from breathing failure. BAG3 normally helps supervise the quality of the tiny contractile units inside muscle fibers, directing damaged proteins to cellular recycling. The authors engineered mice that produce the human mutant form of BAG3 tagged with a fluorescent marker, allowing them to track its behavior. Unlike mice expressing normal human BAG3, animals with the mutant version rapidly became smaller, showed wasting of limb and breathing muscles, and had a waddling gait, closely echoing the human disease.
How Muscle Fibers Fall Apart
Microscopic and ultrastructural examinations revealed how thoroughly the mutant protein disrupts muscle structure. In affected mice, muscle fibers were thinner and riddled with abnormal protein clumps. The highly ordered stripes of the contractile machinery—the sarcomeres—were fragmented and blurred, and many fibers showed centralized nuclei, a hallmark of attempted regeneration. Immune cells infiltrated the tissue and fibrous scar material increased, though less dramatically than in the heart. Functionally, isolated muscles from these mice produced up to 90 percent less force than those from healthy animals, even after accounting for their smaller size, confirming that the core machinery for contraction was failing.
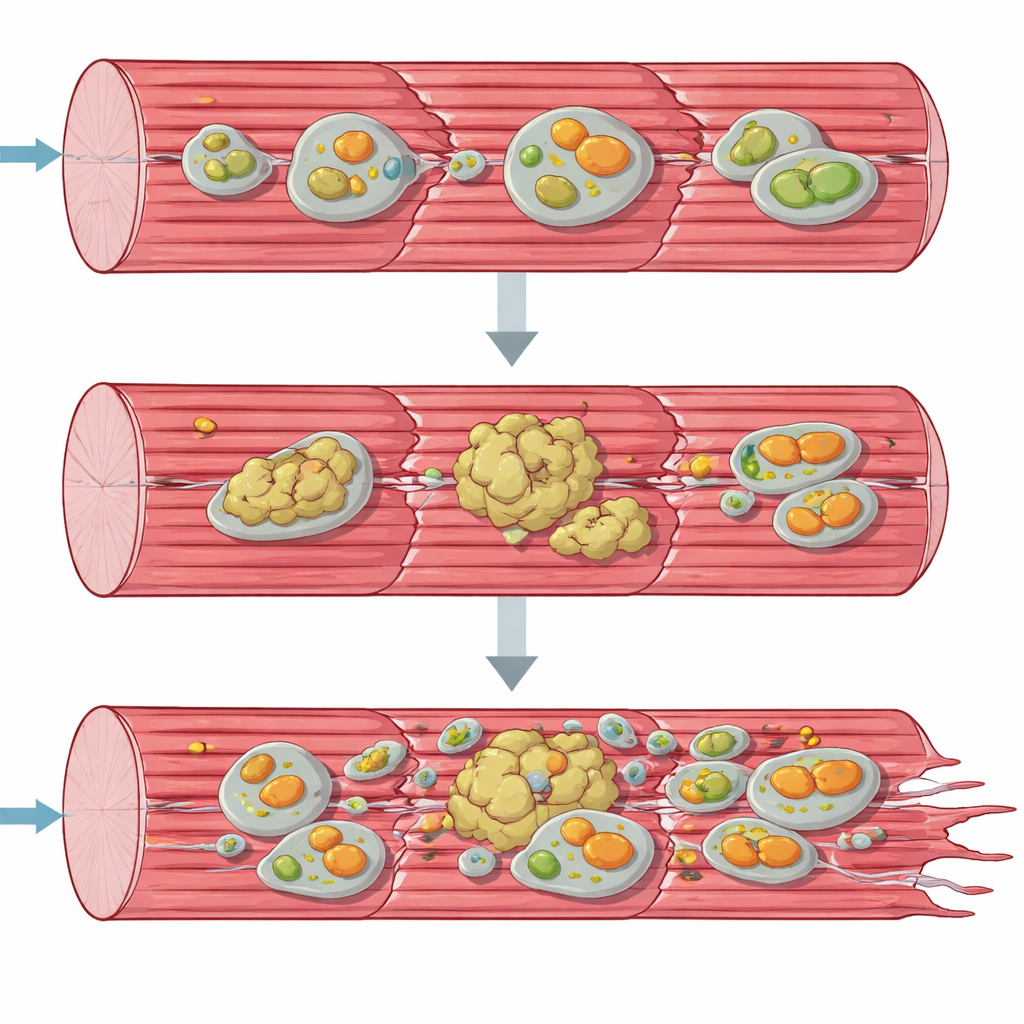
Protein Pileups and Sick Mitochondria
To understand the breakdown from a systems view, the team turned to “omics” tools that survey thousands of molecules at once. Gene activity and protein profiles showed that muscle cells were frantically trying to respond: genes for building new proteins and for stress responses were revved up, and many chaperones and other quality-control factors accumulated. Yet, many key structural proteins of the contractile apparatus were depleted from their normal soluble forms and instead shifted into an insoluble, aggregate-rich fraction. At the same time, components of the cell’s recycling pathways—autophagy and the specialized removal of damaged mitochondria, mitophagy—piled up as if stuck mid-process. Electron microscopy and mitochondrial function tests confirmed that energy-producing organelles were misshapen, clustered, and worked far below normal capacity, especially at the first step of the respiratory chain.
Autophagy as the Central Culprit
The authors then asked whether the primary problem lay in general cellular recycling (autophagy), in mitochondrial-specific recycling (mitophagy), or both. They grew muscle cells from the mutant mice and experimentally boosted different pathways. Stimulating broad autophagy with the protein BECN1 cleared key stress markers, reduced mutant BAG3 aggregates, and improved the appearance and organization of sarcomeres. In contrast, forcing mitophagy or simply increasing mitochondrial production did not correct the defects. In live mice, treatment with the autophagy-activating drug rapamycin improved grip strength, coordination, and movement, and increased markers of active recycling in muscle. Together, these experiments point to a primary block in autophagy, with mitochondrial damage emerging as a secondary consequence rather than the root cause.
Testing a Gene Therapy Strategy
Because the disease is driven by a toxic protein, the researchers explored whether dialing down mutant BAG3 could rescue the muscles. They packaged a short hairpin RNA designed to specifically target human BAG3 into an engineered virus and delivered it into the bloodstream of young mutant mice. Weeks later, muscles showed much less mutant BAG3, fewer protein aggregates, reduced scarring, and far fewer fibers with centralized nuclei. Most importantly, treated muscles generated dramatically more force and partially regained normal size. This proof-of-concept suggests that silencing the disease-causing gene, or combining such an approach with drugs that boost autophagy, could be a powerful strategy for patients, once an allele-specific therapy is optimized.
What This Means for Patients and Beyond
In accessible terms, this work shows that when a key helper protein like BAG3 misfolds and clumps, the cell’s garbage disposal system jams. Broken components and faulty mitochondria accumulate, the muscle’s internal scaffolding collapses, and strength ebbs away. By carefully recreating the disease in mice, the study reveals that restoring or bypassing this cleanup system—either by enhancing autophagy or by reducing mutant BAG3 itself—can substantially restore muscle health. While much remains to be done before such treatments reach the clinic, the findings provide a detailed roadmap for tackling this rare but deadly muscle disorder and offer broader insights into how impaired cellular housekeeping may underlie other degenerative diseases.
Citation: Filippi, K., Graf-Riesen, K., Kuppusamy, M. et al. Blockage of autophagy causes severe skeletal muscle disruption in a mouse model for myofibrillar myopathy 6. Nat Commun 17, 3436 (2026). https://doi.org/10.1038/s41467-026-71749-6
Keywords: myofibrillar myopathy, BAG3 mutation, autophagy, skeletal muscle degeneration, gene therapy