Clear Sky Science · fr
AMR-GNN : un cadre de réseaux de neurones graphiques multi-représentations pour permettre la prédiction génomique de la résistance aux antimicrobiens
Pourquoi il est crucial de prédire la résistance aux médicaments
Les infections résistantes aux antibiotiques figurent parmi les menaces médicales majeures de notre époque, tuant plus d’un million de personnes chaque année. Les médecins ont un besoin urgent de méthodes plus rapides pour savoir quels antibiotiques seront efficaces pour une infection donnée, mais les tests de laboratoire traditionnels peuvent prendre plusieurs jours. Cette étude présente un nouveau cadre d’intelligence artificielle, nommé AMR-GNN, qui lit la séquence d’ADN complète des bactéries et prédit si elles seront résistantes ou sensibles à différents antibiotiques, ouvrant potentiellement la voie à des recommandations le jour même, au chevet du patient.
Des cultures lentes aux tests numériques d’ADN
Aujourd’hui, la plupart des hôpitaux s’appuient encore sur des tests basés sur la culture : les bactéries sont cultivées en laboratoire et exposées à divers médicaments pour voir lesquels arrêtent leur croissance. Bien que fiables, ces méthodes sont lentes et gourmandes en main-d’œuvre. Parallèlement, le séquençage des génomes bactériens entiers est devenu moins cher et plus accessible, générant d’énormes quantités d’informations détaillées. Le défi tient au fait que l’ADN bactérien est extrêmement haute dimension, contenant des millions d’éléments, et il n’existe pas de méthode unique acceptée pour convertir ce code génétique en un format exploitable par des ordinateurs pour prédire la résistance aux médicaments. Les outils antérieurs se concentraient souvent sur quelques gènes de résistance connus ou sur des motifs simples, ce qui fonctionne lorsque la résistance provient d’une seule mutation mais échoue lorsque de nombreuses modifications subtiles interagissent.
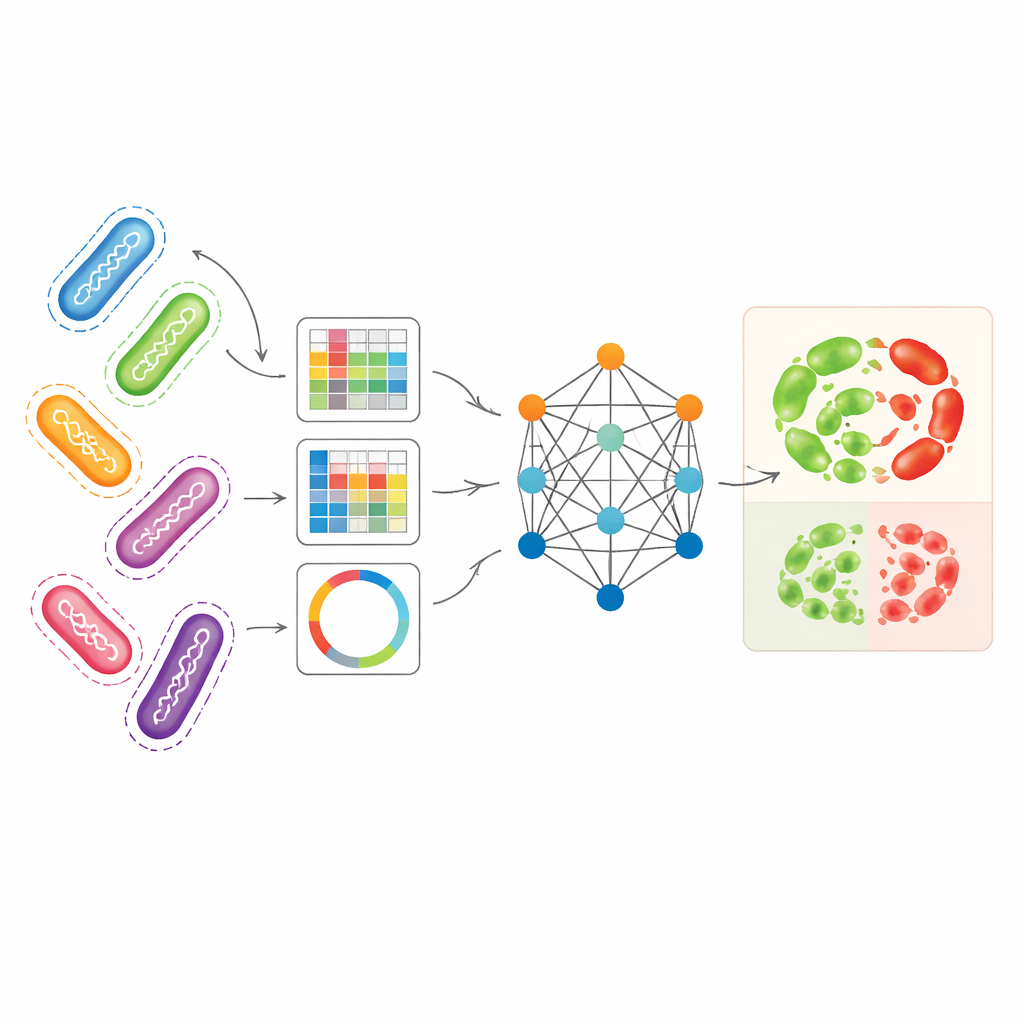
Combiner plusieurs vues génomiques en une seule image
Les chercheurs ont cherché à exploiter plusieurs façons complémentaires de représenter les génomes bactériens simultanément. Ils ont utilisé Pseudomonas aeruginosa — un pathogène acquis en milieu hospitalier aux schémas de résistance particulièrement complexes — comme cas d’étude principal. Une représentation, appelée unitigs, capture des fragments d’ADN récurrents sans dépendre d’un génome de référence. Une autre représentation suit de petites variations d’ADN à des positions spécifiques, tandis qu’une troisième convertit des gènes liés à la résistance en motifs de type image qui résument l’agencement de courts segments d’ADN. Pris isolément, ces types de représentation permettaient déjà à des modèles d’apprentissage automatique classiques de prédire la résistance avec une précision raisonnable, en particulier les unitigs pour certains antibiotiques. Mais chaque vue manque une partie de l’histoire biologique, et les utiliser séparément sous-exploite la richesse des données génomiques sous-jacentes.
Comment le modèle basé sur un graphe apprend à partir de souches apparentées
AMR-GNN utilise une forme d’apprentissage profond connue sous le nom de réseau de neurones graphique, qui traite chaque isolat bactérien comme un point (un nœud) et relie par des arêtes les isolats génétiquement similaires. Dans ce dispositif, le profil détaillé en unitigs de chaque isolat constitue son vecteur de caractéristiques principal, tandis que les autres vues génomiques définissent comment les isolats sont connectés dans le graphe. Le modèle fait ensuite circuler l’information le long de ces connexions, ce qui lui permet d’apprendre à partir des motifs partagés entre génomes apparentés. Pour éviter d’être trompé par de simples relations clonales — où des bactéries sont étroitement apparentées mais diffèrent en résistance pour des raisons que le modèle doit découvrir — les auteurs ont délibérément supprimé les arêtes reliant des isolats appartenant au même groupe de lignée génétique. Cette étape de « découplage » oblige le réseau à prêter davantage attention aux caractéristiques d’ADN spécifiques associées à la résistance, plutôt que de s’appuyer sur des étiquettes de lignée larges comme raccourcis.
Des prédictions renforcées pour plusieurs bactéries et médicaments
Lorsque l’équipe a comparé AMR-GNN à des modèles plus simples reposant sur des vues génomiques uniques, l’approche basée sur les graphes a amélioré les performances pour presque tous les 12 antibiotiques testés chez P. aeruginosa, avec les gains les plus importants pour les médicaments qui étaient auparavant les plus difficiles à prédire. Le modèle s’est également mieux généralisé à des jeux de données de test indépendants, bien que les performances aient encore légèrement diminué en dehors des données d’entraînement, soulignant la nécessité de collections de génomes plus nombreuses et diversifiées. Au-delà de P. aeruginosa, les chercheurs ont appliqué AMR-GNN à plus de 23 000 génomes d’autres agents pathogènes majeurs, notamment Escherichia coli, Klebsiella pneumoniae, Staphylococcus aureus et Enterococcus faecium, sur de nombreux antibiotiques d’importance clinique. Pour la plupart des combinaisons espèce–médicament, le cadre a atteint une très grande précision et a surpassé des outils basés sur des règles largement utilisés, qui dépendent de listes annotées de gènes de résistance connus.
Rendre les modèles boîte noire plus explicables
Une préoccupation importante pour l’usage clinique est de savoir si un tel système d’IA peut fournir un aperçu des raisons d’une prédiction donnée. L’équipe a abordé cette question en appliquant des méthodes d’interprétabilité qui retracent quelles caractéristiques d’ADN contribuent le plus aux décisions du modèle. Pour les médicaments où le modèle a obtenu ses meilleures performances, AMR-GNN a mis en évidence de nombreux gènes et mutations de résistance connus, tels que les cibles classiques des fluoroquinolones. Il a aussi pointé des gènes moins bien compris dont les modifications étaient fortement associées à des concentrations d’antibiotique plus élevées nécessaires pour inhiber la croissance bactérienne, suggérant de nouveaux candidats pour des investigations expérimentales en laboratoire. Cette capacité à prédire la résistance tout en signalant des moteurs biologiques potentiels aide à combler le fossé entre prédiction pure et compréhension mécanistique.
Ce que cela signifie pour les soins aux patients à l’avenir
En substance, ce travail montre que la combinaison de plusieurs « vues » de l’ADN au sein d’un modèle d’apprentissage profond basé sur un graphe peut considérablement améliorer la prédiction de la résistance aux antibiotiques à partir des génomes bactériens. AMR-GNN est présenté comme un cadre flexible et interprétable qui peut être étendu à d’autres types de données, comme des mesures d’activité des gènes ou des informations cliniques. Bien que des travaux supplémentaires soient nécessaires — en particulier des ensembles de données plus importants et géographiquement variés ainsi que des essais cliniques prospectifs — l’approche nous rapproche d’un futur où une séquence génomique bactérienne, obtenue directement à partir d’un prélèvement patient, pourrait rapidement orienter le médecin vers le bon traitement et contribuer à ralentir la propagation des infections résistantes.
Citation: Nguyen, HA., Peleg, A.Y., Wisniewski, J.A. et al. AMR-GNN: a multi-representation graph neural network framework to enable genomic antimicrobial resistance prediction. Nat Commun 17, 3555 (2026). https://doi.org/10.1038/s41467-026-69934-8
Mots-clés: résistance aux antimicrobiens, réseaux de neurones graphiques, génomique bactérienne, apprentissage automatique, prévision de la sensibilité aux antibiotiques