Clear Sky Science · es
AMR-GNN: un marco de redes neuronales de grafos con múltiples representaciones para facilitar la predicción genómica de la resistencia antimicrobiana
Por qué importa predecir la resistencia a los fármacos
Las infecciones resistentes a los antibióticos son una de las principales amenazas médicas de nuestro tiempo, causando más de un millón de muertes cada año. Los médicos necesitan con urgencia formas más rápidas de saber qué antibióticos funcionarán para una infección determinada, pero las pruebas de laboratorio tradicionales pueden tardar varios días. Este estudio presenta un nuevo marco de inteligencia artificial, llamado AMR-GNN, que lee la secuencia completa del ADN bacteriano y predice si serán resistentes o susceptibles a distintos antibióticos, allanando potencialmente el camino para orientar el tratamiento el mismo día junto al paciente.
De los cultivos lentos a las pruebas digitales del ADN
Hoy en día, la mayoría de los hospitales todavía dependen de pruebas basadas en cultivos: las bacterias se cultivan en el laboratorio y se exponen a diversos fármacos para ver cuáles detienen su crecimiento. Aunque fiable, este enfoque es lento y laborioso. Al mismo tiempo, la secuenciación de genomas bacterianos completos se ha abaratado y facilitado, generando enormes cantidades de información detallada. El reto es que el ADN bacteriano es extremadamente de alta dimensión, contiene millones de bloques constructores, y no existe una forma única y acordada de convertir este código genético en un formato que las computadoras puedan usar fácilmente para predecir la resistencia a fármacos. Herramientas anteriores a menudo se centraban en algunos genes de resistencia conocidos o patrones simples, lo que funciona bien cuando la resistencia se debe a una sola mutación pero falla cuando muchas alteraciones sutiles interactúan.
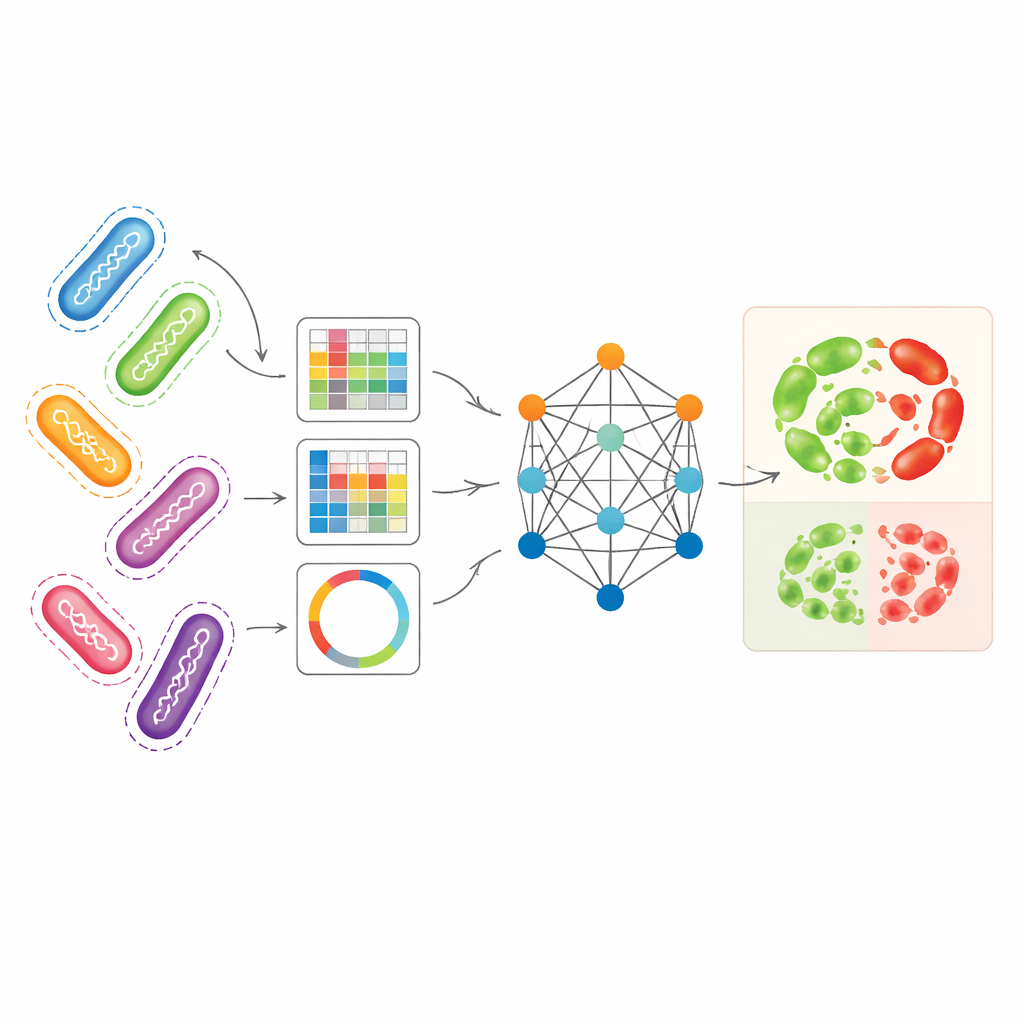
Combinando distintas perspectivas genómicas en una sola imagen
Los investigadores se propusieron aprovechar varias formas complementarias de representar genomas bacterianos a la vez. Usaron Pseudomonas aeruginosa—un patógeno adquirido en hospitales con patrones de resistencia particularmente complejos—como su caso de prueba principal. Una representación, llamada unitigs, captura fragmentos de ADN recurrentes sin depender de un genoma de referencia. Otra representación rastrea pequeños cambios de ADN en posiciones específicas, mientras que una tercera convierte genes relacionados con la resistencia en patrones similares a imágenes que resumen cómo se organizan tramos cortos de ADN. Por sí solas, estas representaciones ya permitían a modelos de aprendizaje automático estándar predecir la resistencia con una precisión razonable, especialmente las unitigs para ciertos antibióticos. Pero cada vista pierde parte de la historia biológica, y usarlas por separado subutiliza la riqueza de los datos genómicos subyacentes.
Cómo el modelo basado en grafos aprende de cepas relacionadas
AMR-GNN utiliza una forma de aprendizaje profundo conocida como red neuronal de grafos, que trata cada aislado bacteriano como un punto (un nodo) y conecta aislados que son genéticamente similares con enlaces (aristas). En esta configuración, el perfil detallado de unitigs de cada aislado constituye su vector de características principal, mientras que las otras vistas genómicas definen cómo se conectan los aislados en el grafo. El modelo luego transmite información a lo largo de estas conexiones, lo que le permite aprender de patrones compartidos entre genomas relacionados. Para evitar ser engañado por relaciones clonales simples —donde las bacterias están estrechamente relacionadas pero difieren en resistencia por razones que el modelo debe descubrir— los autores eliminaron deliberadamente aristas que enlazaban aislados del mismo grupo de línea genética. Este paso de "desacoplamiento" obliga a la red a prestar más atención a las características específicas del ADN asociadas con la resistencia, en lugar de apoyarse en etiquetas de linaje amplias como atajos.
Predicciones más robustas entre bacterias y fármacos
Cuando el equipo comparó AMR-GNN con modelos más simples que se basaban en vistas genómicas individuales, el enfoque basado en grafos mejoró el rendimiento para casi todos los 12 antibióticos probados en P. aeruginosa, con las mayores ganancias en fármacos que antes eran más difíciles de predecir. El modelo también se generalizó mejor a conjuntos de prueba independientes, aunque el rendimiento aún disminuyó algo fuera de los datos de entrenamiento, lo que subraya la necesidad de colecciones de genomas más grandes y diversas. Más allá de P. aeruginosa, los investigadores aplicaron AMR-GNN a más de 23.000 genomas de otros patógenos importantes, incluidos Escherichia coli, Klebsiella pneumoniae, Staphylococcus aureus y Enterococcus faecium, frente a muchos antibióticos clínicamente relevantes. En la mayoría de las combinaciones especie–fármaco, el marco alcanzó una precisión muy alta y superó a herramientas basadas en reglas ampliamente usadas que dependen de listas curadas de genes de resistencia conocidos.
Haciendo los modelos de caja negra más explicables
Una preocupación importante para el uso clínico es si un sistema de IA así puede ofrecer información sobre por qué hace una predicción concreta. El equipo abordó esto aplicando métodos de interpretabilidad que rastrean qué características del ADN contribuyen más a las decisiones del modelo. Para los fármacos donde el modelo funcionó mejor, AMR-GNN destacó muchos genes y mutaciones de resistencia conocidos, como dianas clásicas de los antibióticos fluoroquinolónicos. También señaló genes menos comprendidos cuyos cambios se asociaron fuertemente con concentraciones más altas de fármaco necesarias para detener el crecimiento bacteriano, lo que sugiere nuevos candidatos para estudios experimentales. Esta capacidad de predecir la resistencia y al mismo tiempo señalar posibles impulsores biológicos ayuda a cerrar la brecha entre la pura predicción y la comprensión mecanicista.
Qué significa esto para la atención futura al paciente
En esencia, este trabajo muestra que combinar múltiples "vistas" del ADN dentro de un modelo de aprendizaje profundo basado en grafos puede mejorar sustancialmente la predicción de la resistencia a antibióticos a partir de genomas bacterianos. AMR-GNN se presenta como un marco flexible y interpretable que puede ampliarse a otros tipos de datos, como mediciones de actividad génica o información clínica. Aunque se necesita más trabajo—especialmente conjuntos de datos más grandes, geográficamente variados y ensayos clínicos prospectivos—el enfoque nos acerca a un futuro en el que la secuencia del genoma de una bacteria, obtenida directamente de una muestra de paciente, podría guiar rápidamente a los médicos hacia el fármaco adecuado y ayudar a frenar la propagación de infecciones resistentes.
Cita: Nguyen, HA., Peleg, A.Y., Wisniewski, J.A. et al. AMR-GNN: a multi-representation graph neural network framework to enable genomic antimicrobial resistance prediction. Nat Commun 17, 3555 (2026). https://doi.org/10.1038/s41467-026-69934-8
Palabras clave: resistencia antimicrobiana, redes neuronales de grafos, genómica bacteriana, aprendizaje automático, predicción de susceptibilidad a antibióticos