Clear Sky Science · de
AMR-GNN: ein Multi-Representation-Graph-Neural-Network-Framework zur Vorhersage genomischer antimikrobieller Resistenz
Warum die Vorhersage von Arzneimittelresistenzen wichtig ist
Antibiotikaresistente Infektionen gehören zu den größten gesundheitlichen Bedrohungen unserer Zeit und töten jährlich mehr als eine Million Menschen. Ärztinnen und Ärzte benötigen dringend schnellere Methoden, um festzustellen, welche Antibiotika bei einer bestimmten Infektion wirken, doch traditionelle Labortests können mehrere Tage dauern. Diese Studie stellt ein neues Rahmenwerk der künstlichen Intelligenz vor, genannt AMR-GNN, das die vollständige DNA-Sequenz von Bakterien analysiert und vorhersagt, ob sie gegenüber verschiedenen Antibiotika resistent oder empfindlich sind — womöglich ein Schritt hin zu tagesgleichen Therapieempfehlungen am Krankenbett.
Von langsamen Kulturen zu digitalen DNA-Tests
Heute verlassen sich die meisten Krankenhäuser noch auf kulturbasierte Tests: Bakterien werden im Labor angezüchtet und verschiedenen Wirkstoffen ausgesetzt, um zu sehen, welche ihr Wachstum stoppen. Zwar zuverlässig, ist dieses Vorgehen jedoch langsam und arbeitsintensiv. Gleichzeitig sind Ganzgenomsequenzierungen von Bakterien günstiger und einfacher geworden und liefern enorme Mengen detaillierter Informationen. Die Herausforderung besteht darin, dass bakterielle DNA extrem hochdimensional ist — sie enthält Millionen Bausteine — und es keine einheitliche Methode gibt, diesen genetischen Code so aufzubereiten, dass Computer ihn leicht zur Vorhersage von Arzneimittelresistenz nutzen können. Frühere Werkzeuge konzentrierten sich häufig auf einige bekannte Resistenzgene oder einfache Muster; das funktioniert gut, wenn die Resistenz durch eine einzelne Mutation verursacht wird, versagt aber, wenn viele subtile Veränderungen miteinander interagieren.
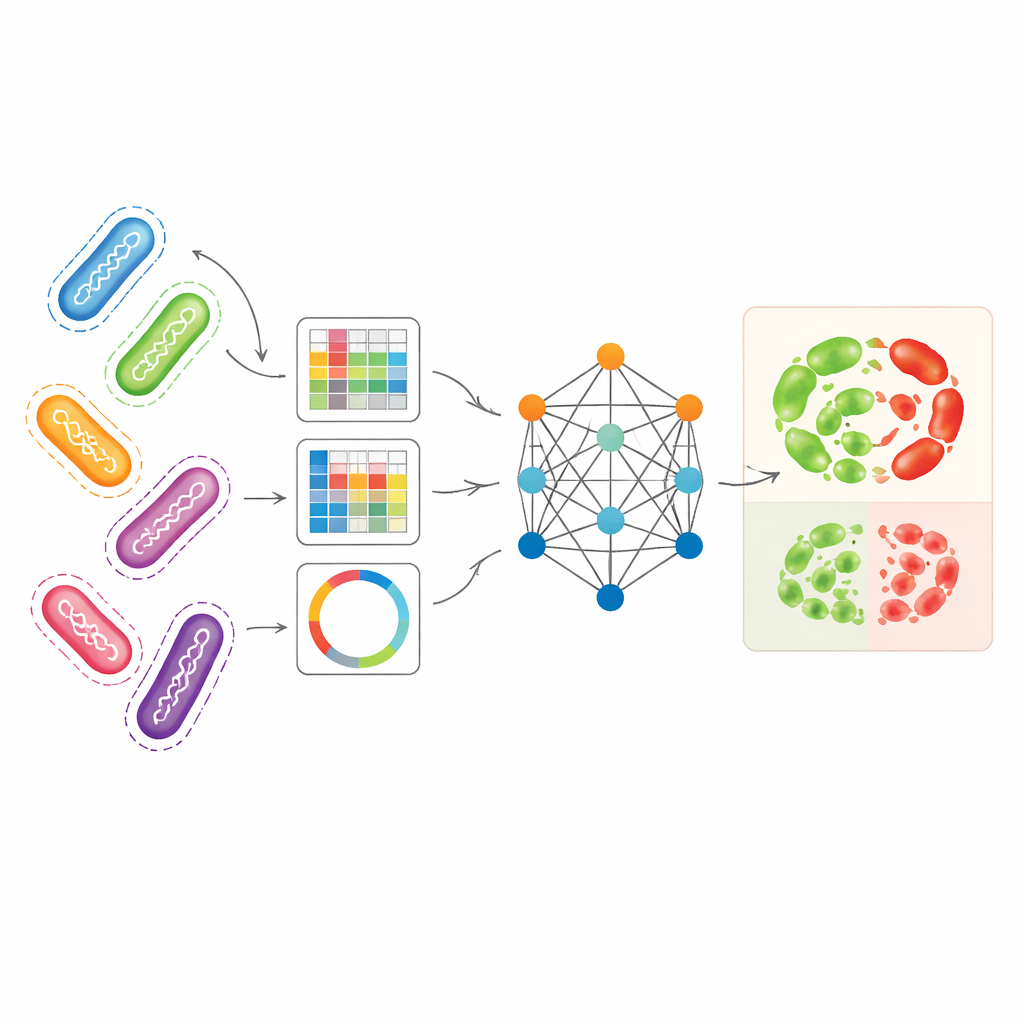
Verschiedene genomische Perspektiven zu einem Bild vereinen
Die Forschenden verfolgten den Ansatz, mehrere komplementäre Darstellungsformen bakterieller Genome gleichzeitig zu nutzen. Als Hauptfallstudie verwendeten sie Pseudomonas aeruginosa — einen im Krankenhaus erworbenen Erreger mit besonders komplexen Resistenzmustern. Eine Darstellung, sogenannte Unitigs, erfasst wiederkehrende DNA-Fragmente, ohne auf ein Referenzgenom angewiesen zu sein. Eine andere Darstellung verfolgt kleine DNA-Veränderungen an spezifischen Positionen, während eine dritte ausgewählte, resistenzrelevante Gene in bildähnliche Muster umwandelt, die zusammenfassen, wie kurze DNA-Abschnitte angeordnet sind. Für sich genommen erlaubten diese Repräsentationen bereits, mit Standardmethoden des maschinellen Lernens die Resistenz mit anständiger Genauigkeit vorherzusagen — insbesondere Unitigs für bestimmte Antibiotika. Jede Sichtweise allein lässt jedoch Teile der biologischen Geschichte aus und nutzt die Fülle der zugrundeliegenden Genomdaten nicht vollständig aus.
Wie das graphbasierte Modell von verwandten Stämmen lernt
AMR-GNN verwendet eine Form des Deep Learning, die als graphneuronales Netzwerk bekannt ist. Dabei wird jede bakterielle Isolatform als Punkt (Knoten) behandelt und Isolate, die genetisch ähnlich sind, werden durch Verbindungen (Kanten) verknüpft. In diesem Aufbau bildet das detaillierte Unitig-Profil jedes Isolats seinen Hauptmerkmalsvektor, während die anderen genomischen Sichten definieren, wie Isolate im Graphen verbunden sind. Das Modell leitet dann Informationen entlang dieser Verbindungen weiter, sodass es aus Mustern lernen kann, die bei verwandten Genomen geteilt werden. Um nicht von einfachen klonalen Beziehungen in die Irre geführt zu werden — also von Fällen, in denen Bakterien eng verwandt sind, sich aber aus Gründen, die das Modell entdecken soll, in der Resistenz unterscheiden — entfernten die Autorinnen und Autoren gezielt Kanten, die Isolate derselben genetischen Liniengruppe verbanden. Dieser Schritt der "Entkopplung" zwingt das Netzwerk, stärkeren Wert auf die spezifischen DNA-Merkmale zu legen, die mit Resistenz assoziiert sind, anstatt auf breite Linienlabels als Abkürzung zurückzugreifen.
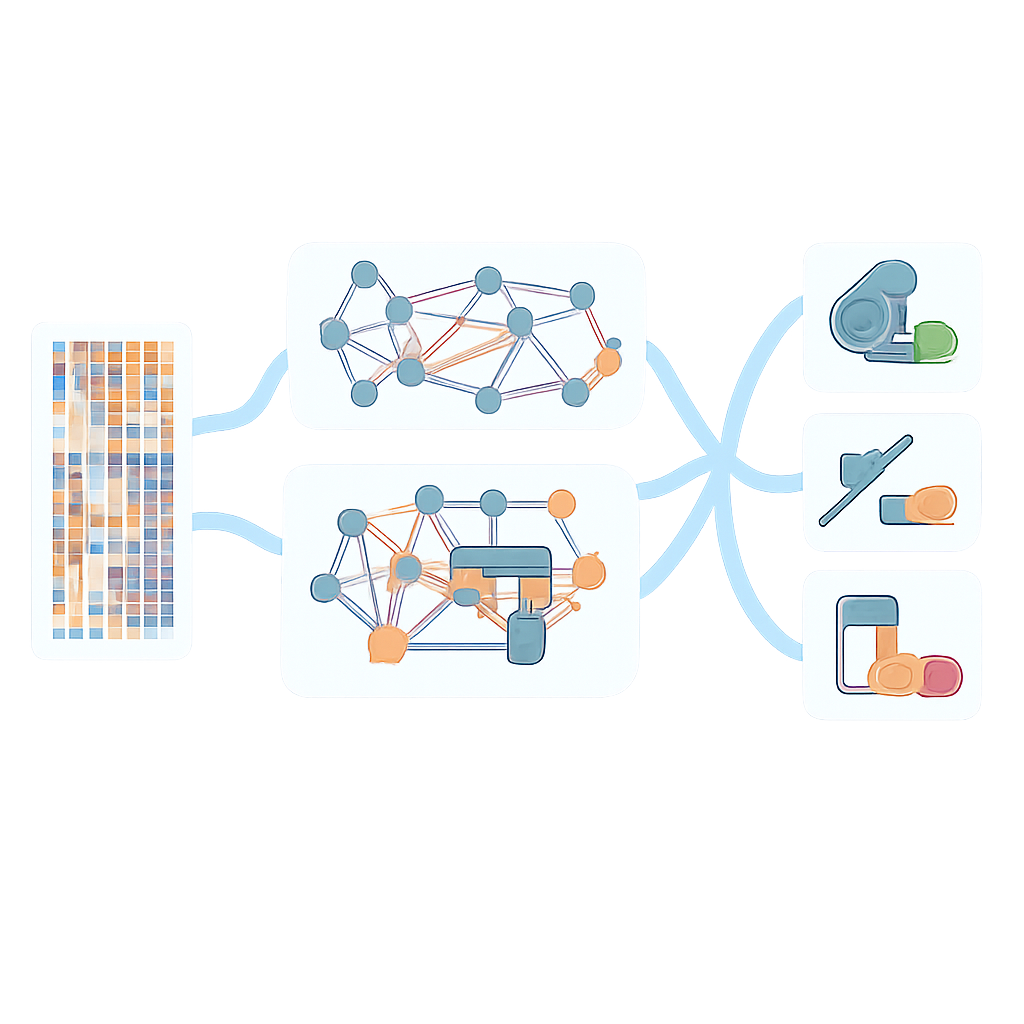
Stärkere Vorhersagen über Bakterien und Wirkstoffe hinweg
Im Vergleich zu einfacheren Modellen, die sich auf einzelne genomische Sichten stützten, verbesserte der graphbasierte Ansatz die Leistung für nahezu alle der 12 in P. aeruginosa getesteten Antibiotika, wobei die größten Gewinne bei den zuvor am schwierigsten vorherzusagenden Wirkstoffen auftraten. Das Modell verallgemeinerte außerdem besser auf unabhängige Testdatensätze, wenngleich die Leistung außerhalb der Trainingsdaten teilweise nachließ — ein Hinweis auf die Notwendigkeit größerer und vielfältigerer Genom-Sammlungen. Über P. aeruginosa hinaus wandten die Forschenden AMR-GNN auf mehr als 23.000 Genome anderer wichtiger Erreger an, darunter Escherichia coli, Klebsiella pneumoniae, Staphylococcus aureus und Enterococcus faecium, für zahlreiche klinisch relevante Antibiotika. In den meisten Spezies–Wirkstoff-Kombinationen erzielte das Framework sehr hohe Genauigkeit und übertraf weit verbreitete regelbasierte Werkzeuge, die auf kuratierten Listen bekannter Resistenzgene beruhen.
Schwarzkästchen-Modelle erklärbarer machen
Eine wichtige Frage für den klinischen Einsatz ist, ob ein solches KI-System Einblick darin geben kann, warum es eine bestimmte Vorhersage trifft. Das Team ging dieses Thema an, indem es Interpretierbarkeitsmethoden anwandte, die nachverfolgen, welche DNA-Merkmale am stärksten zu den Entscheidungen des Modells beitragen. Für die Wirkstoffe, bei denen das Modell am besten abschnitt, hob AMR-GNN viele bekannte Resistenzgene und Mutationen hervor, etwa klassische Ziele von Fluorchinolon-Antibiotika. Es identifizierte auch weniger gut verstandene Gene, deren Veränderungen stark mit höheren Wirkstoffkonzentrationen verbunden waren, die erforderlich sind, um das bakterielle Wachstum zu hemmen — und lieferte damit neue Kandidaten für experimentelle Nachuntersuchungen im Labor. Die Fähigkeit, sowohl Resistenz vorherzusagen als auch potenzielle biologische Treiber zu markieren, hilft, die Lücke zwischen reiner Vorhersage und mechanistischem Verständnis zu schließen.
Was das für die zukünftige Patientenversorgung bedeutet
Im Kern zeigt diese Arbeit, dass die Kombination mehrerer DNA‑"Sichten" in einem graphbasierten Deep‑Learning‑Modell die Vorhersage von Antibiotikaresistenz aus bakteriellen Genomen deutlich verbessern kann. AMR-GNN wird als flexibles, erklärbares Framework präsentiert, das auf andere Datentypen erweiterbar ist, etwa Genaktivitätsmessungen oder klinische Informationen. Zwar sind weitere Arbeiten nötig — insbesondere größere, geografisch breit gefächerte Datensätze und prospektive klinische Studien —, doch rückt der Ansatz uns näher an eine Zukunft, in der eine bakterielle Genomsequenz, direkt aus einer Patientenprobe gewonnen, Ärzten rasch zur richtigen Therapie verhelfen und die Ausbreitung resistenter Infektionen verlangsamen könnte.
Zitation: Nguyen, HA., Peleg, A.Y., Wisniewski, J.A. et al. AMR-GNN: a multi-representation graph neural network framework to enable genomic antimicrobial resistance prediction. Nat Commun 17, 3555 (2026). https://doi.org/10.1038/s41467-026-69934-8
Schlüsselwörter: antimikrobielle Resistenz, Graph-Neuronale Netzwerke, bakterielle Genomik, maschinelles Lernen, Vorhersage der Antibiotikaempfindlichkeit