Clear Sky Science · fr
Flux de travail protéomique multi‑organes à haut débit pour l’analyse de l’efficacité et de la toxicité des médicaments
Pourquoi des cartographies protéiques plus rapides comptent pour la médecine
Nos corps sont composés de milliers de protéines différentes qui évoluent en réponse aux maladies et aux médicaments. Mesurer ces changements simultanément dans plusieurs organes est essentiel pour comprendre pourquoi un traitement fonctionne, quand il échoue et quels effets secondaires il peut provoquer. Cette étude présente une méthode rapide pour quantifier les protéines dans des centaines d’échantillons de tissus par jour et l’applique à un médicament anticancéreux appelé L‑asparaginase, révélant comment ce traitement remodèle la biologie à l’échelle du corps.
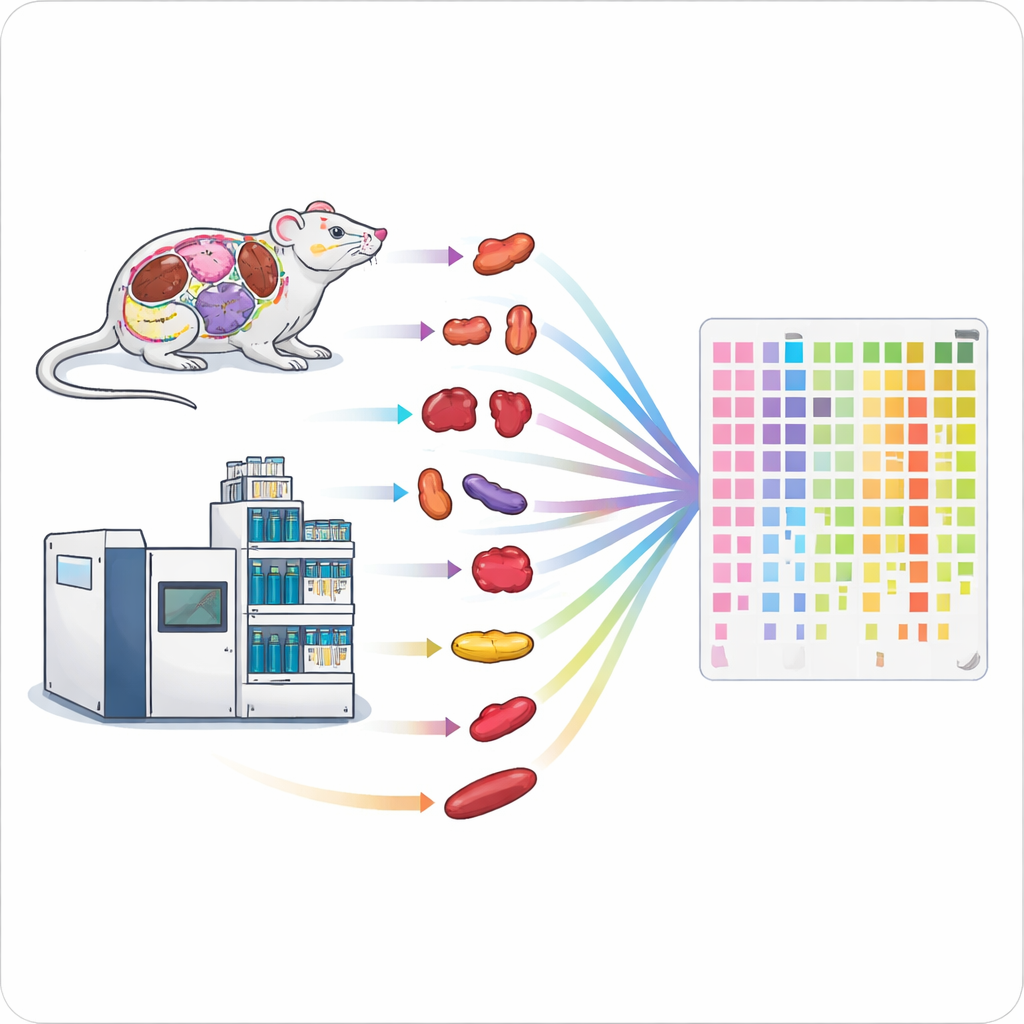
Accélérer la chaîne de production des protéines
Les méthodes traditionnelles de mesure des protéines sont lentes et exigeantes en main‑d’œuvre, ce qui complique l’étude d’organismes entiers ou de larges cohortes de patients. Les auteurs ont combiné deux technologies haute performance — un type rapide de spectrométrie de masse et des cycles de chromatographie en phase liquide courts — en un flux de travail intégré. En ajustant finement des paramètres tels que le temps de collecte des ions et les plages de masse analysées, ils ont obtenu une couverture protéique approfondie en seulement une à deux minutes de mesure par échantillon. Leur configuration optimisée a permis d’identifier environ 6 200 protéines humaines en une minute et près de 7 500 en deux minutes, dépassant largement les méthodes haut débit antérieures.
Du tissu aux données en une seule journée
Collecter rapidement des données ne suffit pas ; la préparation des échantillons peut être un goulot d’étranglement encore plus important. Pour y remédier, l’équipe a conçu une chaîne de traitement parallèle pour tissus solides. Ils ont utilisé des homogénéisateurs automatiques à billes pour lyser jusqu’à 24 morceaux de tissu simultanément, suivis par une digestion des protéines assistée par ultrasons en plaques 96 puits et un désalage rapide à l’aide d’embouts spécialisés. Cette chaîne à haut débit a permis de traiter 96 tissus en environ cinq heures, contre plusieurs jours avec un protocole standard. Des tests sur cerveau, rein et poumon de souris ont montré que la méthode rapide récupérait des nombres de protéines comparables et fournissait des résultats quantitatifs très similaires, mais avec un débit plus de sept fois supérieur.
Observer l’action d’un médicament anticancéreux dans tout le corps
Pour démontrer le potentiel de la plateforme, les chercheurs se sont intéressés à la L‑asparaginase, une enzyme médicamenteuse utilisée contre la leucémie infantile et difficile à réutiliser chez l’adulte en raison d’effets indésirables et de résistances. Ils ont traité des souris avec la L‑asparaginase standard, une version modifiée à activité réduite sur un acide aminé apparenté, ou une solution témoin, puis ont prélevé 13 organes différents sur une période de huit jours. Au total, ils ont quantifié plus de 11 000 protéines sur 507 échantillons. Les schémas dans les données protéiques ont clairement séparé des tissus tels que le cerveau, le foie et les dépôts adipeux, et ont montré que le médicament standard, mais pas la version mutante, entraînait d’importantes variations des niveaux protéiques dans de nombreux organes. Des mesures de petites molécules dans le sang et les tissus ont confirmé que le médicament standard épuisait fortement l’asparagine, tandis que la version mutante n’induisait que des changements brefs, expliquant en partie son efficacité réduite.
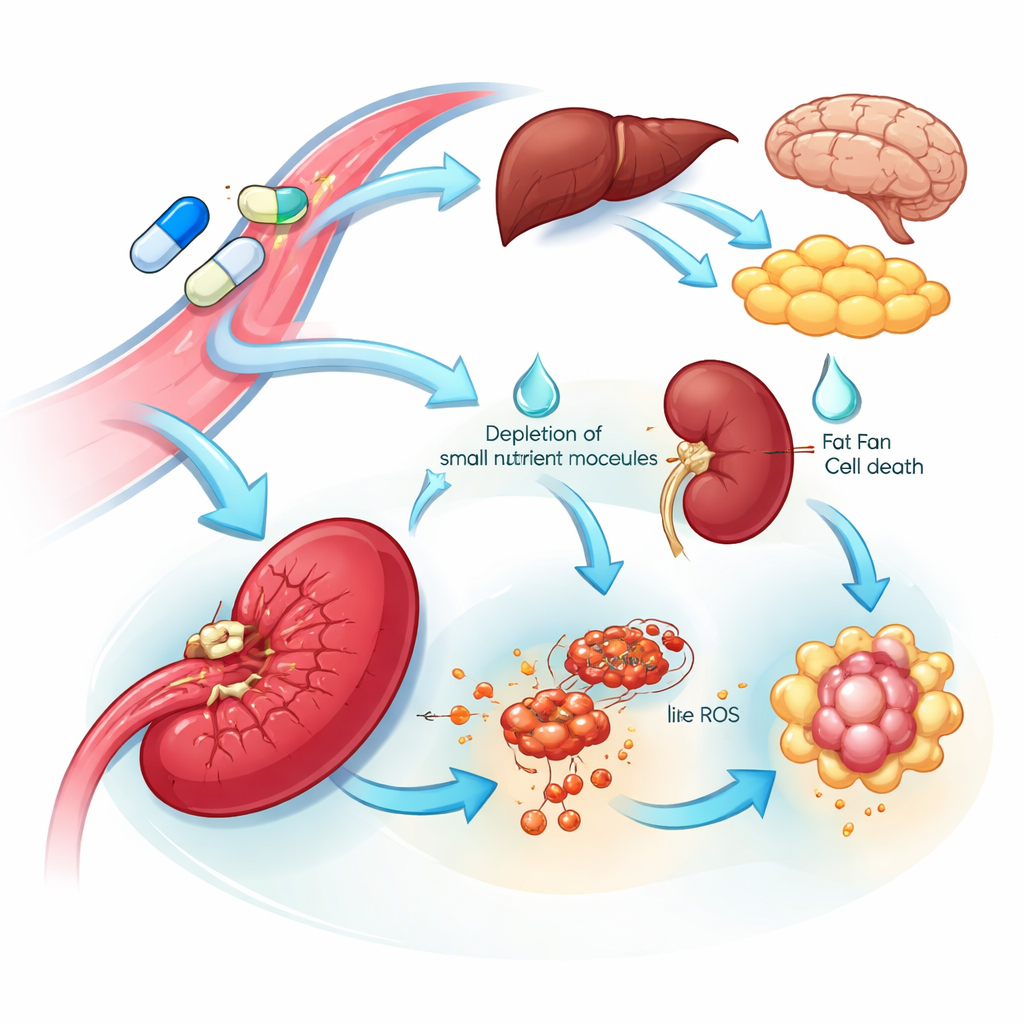
Indices sur la vulnérabilité des organes et les effets secondaires
La vue multi‑organes a mis au jour à la fois des cibles vulnérables et des sources potentielles de toxicité. De nombreux tissus ont répondu à la L‑asparaginase en augmentant l’enzyme qui synthétise l’asparagine, suggérant un programme de résistance intrinsèque visant à restaurer ce nutriment. En revanche, la rate présentait un effondrement net de cette enzyme ainsi qu’une baisse d’acides aminés associés et de précurseurs d’ADN, accompagnés de signes compatibles avec une mort cellulaire induite par le fer. Ces changements indiquent que les cellules liées à la rate pourraient être particulièrement sensibles au médicament. Dans presque tous les organes, le médicament standard — mais pas la version mutante — a supprimé des protéines impliquées dans la régulation de la coagulation sanguine et le métabolisme du cholestérol. Ce ralentissement coordonné et étendu reflète des problèmes cliniques connus du médicament, notamment des troubles de la coagulation et un taux élevé de triglycérides sanguins, et met en évidence des protéines plasmatiques spécifiques qui pourraient être surveillées ou ciblées pour réduire ces risques.
Ce que cela signifie pour les traitements futurs
En associant une plateforme de mesure protéique rapide et évolutive à une étude pharmacologique à l’échelle du corps entier, les auteurs ont créé une sorte d’atlas moléculaire de la L‑asparaginase : où elle agit, comment les organes s’adaptent ou échouent à s’adapter, et quelles voies relient ses bénéfices à ses effets nocifs. Pour un public non spécialiste, le message essentiel est qu’il est désormais possible de scanner des milliers de protéines dans de nombreux organes en une seule journée, transformant des effets médicamenteux complexes en cartes détaillées et testables. De tels flux de travail pourraient accélérer la conception de thérapies anticancéreuses plus sûres, guider les décisions de dose et d’ordonnancement, et aider à identifier des traitements combinés qui conservent le pouvoir anticancéreux tout en atténuant les effets secondaires dangereux.
Citation: Xiong, Y., Tan, L., Chan, Wk. et al. High-throughput multi-organ proteomics workflow for drug efficacy and toxicity analysis. Nat Commun 17, 3505 (2026). https://doi.org/10.1038/s41467-026-69471-4
Mots-clés: protéomique, toxicité des médicaments, thérapie contre le cancer, spectrométrie de masse, biologie des systèmes