Clear Sky Science · fr
La dysfonction de α2δ4 entraîne la dégénérescence des photorécepteurs via la perturbation des mitochondries synaptiques et du dialogue calcique
Pourquoi cette recherche sur l’œil est importante
La perte de vision due à la dégénérescence rétinienne est généralement permanente, et les traitements capables de protéger réellement les cellules sensibles à la lumière de l’œil restent rares. Cette étude pose une question fondamentale : comment de petits problèmes aux jonctions entre cellules de la rétine peuvent-ils à la longue provoquer la mort de ces cellules et, en fin de compte, la cécité ? En suivant un défaut génétique spécifique retrouvé chez certaines personnes atteintes de troubles visuels héréditaires, les auteurs dévoilent un coupable inattendu — de petites centrales énergétiques situées aux terminaisons nerveuses des photorécepteurs — et tracent la manière dont des signaux calciques perturbés aux synapses peuvent progressivement compromettre la santé de la cellule entière.
De la mauvaise vision nocturne aux capteurs de la lumière en voie de disparition
Le travail se concentre sur les photorécepteurs, bâtonnets et cônes qui transforment la lumière en signaux électriques. À leur base, ces cellules forment des synapses spécialisées qui libèrent en continu des signaux chimiques vers les neurones en aval. Une protéine appelée α2δ4 aide à assembler et stabiliser un complexe de canaux calciques (Cav1.4) à ces synapses, et des mutations de son gène ont été associées à des dystrophies rétiniennes humaines. Les patients présentent souvent des problèmes précoces de transmission des signaux des photorécepteurs aux cellules bipolaires, sans perdre un grand nombre de photorécepteurs pendant de nombreuses années. En utilisant des souris génétiquement modifiées dépourvues d’α2δ4, les chercheurs ont cherché à comprendre comment un défaut synaptique apparemment « local » conduit finalement à l’amincissement progressif de la couche contenant les corps cellulaires des photorécepteurs.
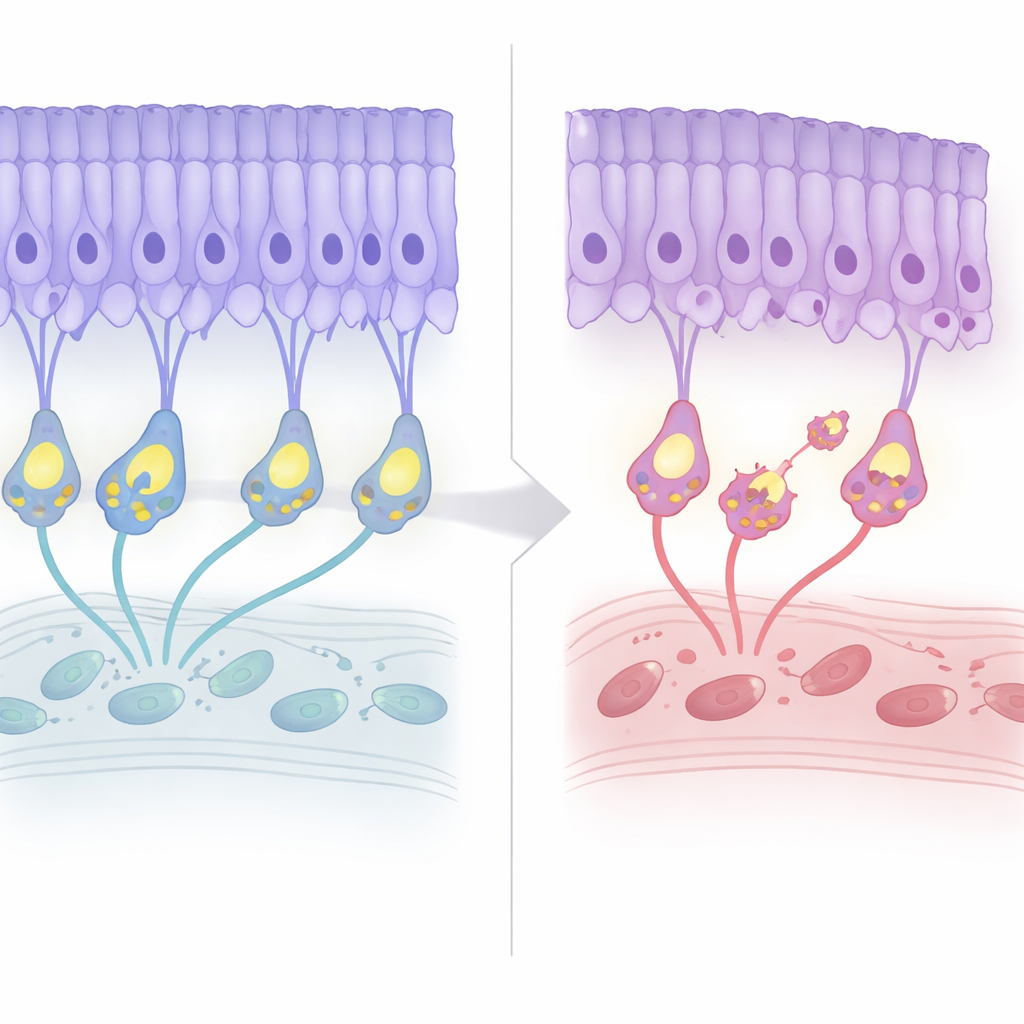
Déclin structurel lent avec signes d’alerte précoces
L’équipe a suivi les rétines de ces souris tout au long de leur vie. Pendant les premiers mois, l’architecture globale semblait normale et les enregistrements électriques suggéraient que la détection de la lumière restait largement intacte. Vers l’âge de sept mois, toutefois, la couche des noyaux des photorécepteurs a commencé à s’amincir d’environ 20 % puis s’est stabilisée, reproduisant la dégénérescence légère et d’apparition tardive observée chez les patients. Fait intéressant, des marqueurs de mort cellulaire programmée sont apparus plus tôt, avant une perte cellulaire évidente. Parallèlement, le « câblage » s’est réorganisé : les terminaisons des photorécepteurs se sont retirées de leur couche habituelle, et les dendrites des cellules bipolaires ont poussé vers le haut pour les rejoindre, formant des connexions ectopiques. Malgré ce remodelage, l’efficacité de la transmission synaptique — évaluée par le ratio des signaux électriques en aval par rapport à ceux en amont — est demeurée constamment faible mais ne s’est pas aggravée avec l’âge.
Les centrales énergétiques synaptiques sous stress
Pour explorer ce qui se passe avant la mort cellulaire, les auteurs ont comparé la composition protéique des rétines jeunes mutantes et normales. Le changement précoce le plus marquant fut une baisse étendue des composants du cycle de l’acide citrique (cycle TCA), une voie centrale de production d’énergie. La microscopie a confirmé que les mitochondries, organites hébergeant ce cycle, étaient particulièrement touchées au niveau des synapses des photorécepteurs. Chez les souris saines, ces terminaisons contiennent des mitochondries proéminentes ; chez les animaux déficients en α2δ4, les mitochondries synaptiques étaient nettement plus petites et moins nombreuses, en particulier dans les terminaisons des cônes, et ce bien avant une dégénérescence généralisée. Les marqueurs mitochondriaux dans la couche synaptique ont diminué, et certaines mitochondries semblaient déplacées vers la couche des noyaux des photorécepteurs. En revanche, les mitochondries du segment interne — le principal centre métabolique du photorécepteur — étaient relativement préservées au début, soulignant une vulnérabilité particulière des mitochondries synaptiques.
Conversations calciques rompues entre compartiments cellulaires
Parce qu’α2δ4 fait partie d’un complexe de canaux calciques, l’équipe a testé si une perturbation de la gestion du calcium pouvait être à l’origine des problèmes mitochondriaux. Dans les photorécepteurs sains, le calcium entrant par les canaux Cav1.4 à la synapse est étroitement coordonné avec le stockage et la libération de calcium par le réticulum endoplasmique (RE) et l’absorption par les mitochondries, créant un microcircuit qui relie l’activité électrique à l’approvisionnement énergétique. Chez les souris mutantes, des protéines libérant le calcium du RE (comme le récepteur à la ryanodine RYR2) et un transporteur mitochondrial clé du calcium (MCU) étaient spécifiquement réduits dans la couche synaptique. Les marqueurs de stress du RE étaient élevés, suggérant que ce compartiment était lui aussi soumis à une tension. Ensemble, ces changements indiquent une rupture du « dialogue » calcique local entre la membrane cellulaire, le RE et les mitochondries là où se produisent les signaux synaptiques.
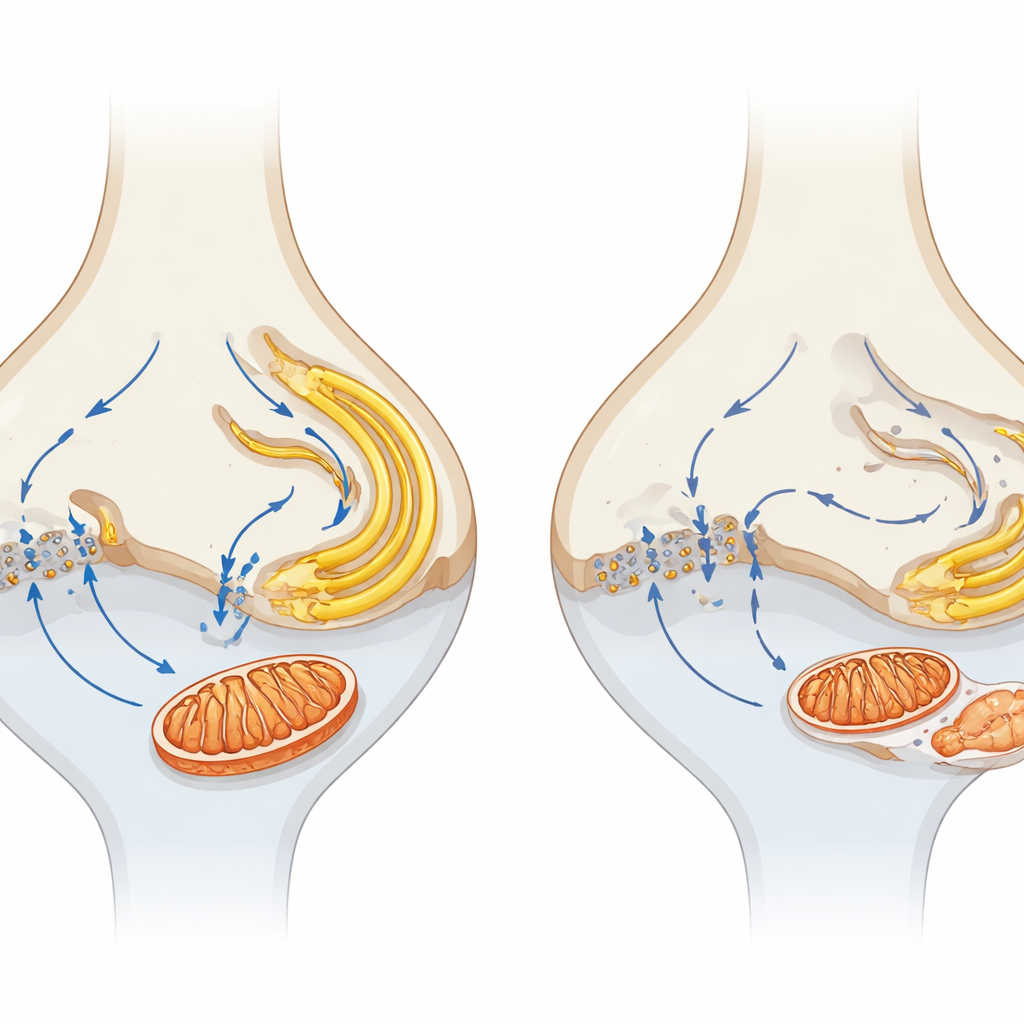
Relier les petites synapses à la perte de vision à long terme
En comparant des souris knock-out pour α2δ4 avec des animaux dépourvus complètement du canal Cav1.4, les auteurs ont constaté qu’une perturbation plus sévère du canal entraîne une dégénérescence plus rapide et plus prononcée, renforçant le rôle central de l’entrée calcique. Ils proposent un modèle dans lequel la perte d’α2δ4 affaiblit la fonction de Cav1.4, perturbe le flux calcique à la synapse et le découple du RE et des mitochondries voisines. Avec le temps, cela conduit à des mitochondries synaptiques endommagées, une production d’énergie altérée et un stress des membranes internes, qui ensemble préparent les photorécepteurs à une mort lente et progressive. Pour le lecteur non spécialisé, l’idée clé est que la vision dépend non seulement de la présence de photorécepteurs, mais aussi du maintien en bon état de leurs petites centrales synaptiques et de leurs signaux calciques ; cibler ces changements locaux et précoces pourrait offrir de nouvelles stratégies pour préserver la vue dans un large éventail de maladies rétiniennes.
Citation: Amieghemen, C.I., Ung, T.T., Huskin, G.N. et al. Dysfunction of α2δ4 leads to photoreceptor degeneration through disrupted synaptic mitochondria and calcium crosstalk. Cell Death Dis 17, 337 (2026). https://doi.org/10.1038/s41419-026-08587-3
Mots-clés: dégénérescence rétinienne, photorécepteurs, mitochondries, signalisation calcique, dysfonction synaptique