Clear Sky Science · en
Dysfunction of α2δ4 leads to photoreceptor degeneration through disrupted synaptic mitochondria and calcium crosstalk
Why this eye research matters
Vision loss from retinal degeneration is usually permanent, and treatments that truly protect the light-sensing cells of the eye remain scarce. This study asks a fundamental question: how can tiny problems at the connections between cells in the retina slowly snowball into the death of those cells and, ultimately, blindness? By following a specific genetic defect found in some people with inherited vision disorders, the authors reveal an unexpected culprit—small energy factories at the nerve endings of photoreceptors—and trace how disturbed calcium signals at synapses can gradually undermine the health of the entire cell.
From faulty night vision to dying light sensors
The work centers on photoreceptors, the rods and cones that convert light into electrical signals. At their base, these cells form specialized synapses that continuously release chemical signals to downstream neurons. A protein called α2δ4 helps assemble and stabilize a calcium channel complex (Cav1.4) at these synapses, and mutations in its gene have been linked to human retinal dystrophies. Patients often show early problems transmitting signals from photoreceptors to bipolar cells but may not lose large numbers of photoreceptors for many years. Using mice engineered to lack α2δ4, the researchers set out to understand how an apparently “local” synaptic defect eventually leads to the slow thinning of the layer that contains photoreceptor cell bodies.
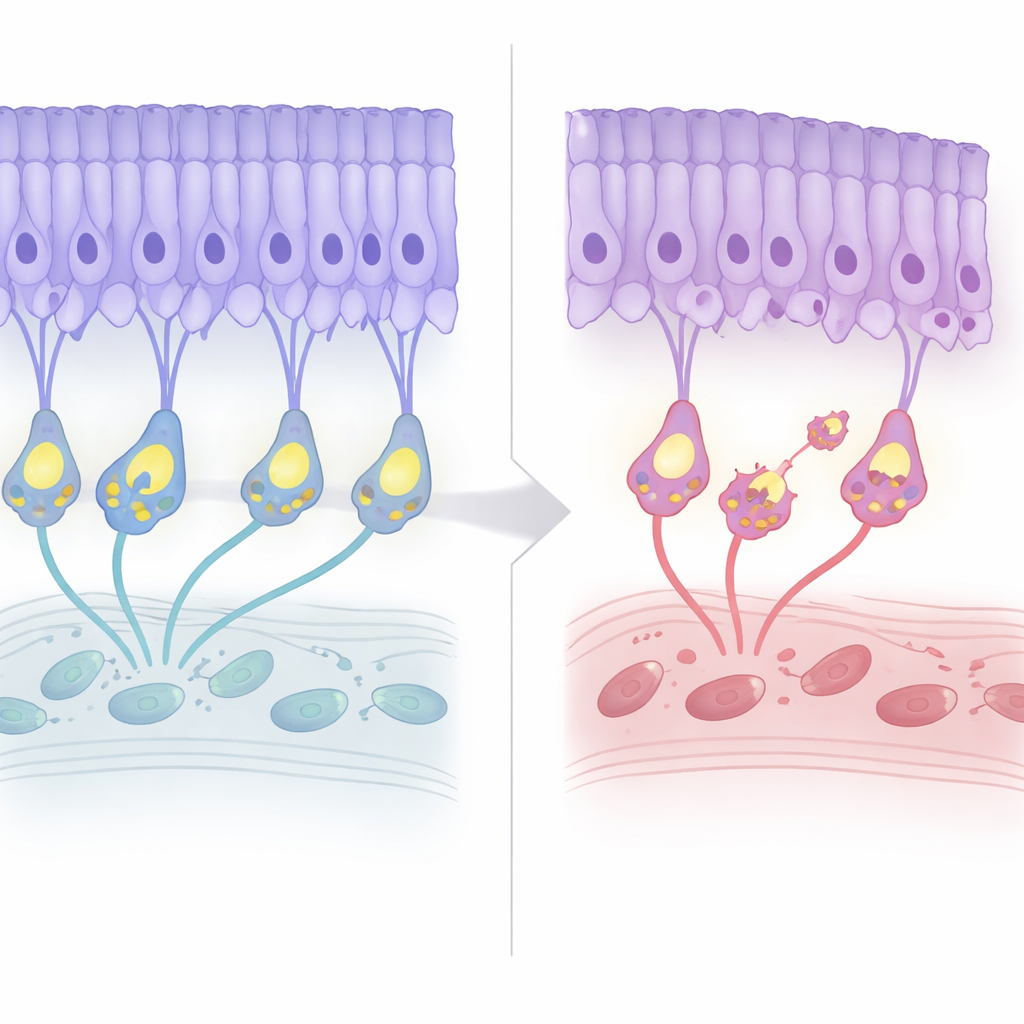
Slow structural decline with early warning signs
The team followed the retinas of these mice across their lifespan. For the first few months, the overall architecture looked normal, and electrical recordings suggested that light detection itself remained largely intact. Around seven months of age, however, the layer of photoreceptor nuclei began to thin by about 20 percent and then plateaued, mirroring the mild, late-onset degeneration seen in patients. Interestingly, markers of programmed cell death appeared earlier, before obvious cell loss. At the same time, the “wiring” reorganized: photoreceptor terminals pulled back from their usual layer, and the dendrites of bipolar cells sprouted upward to meet them, forming ectopic connections. Despite this remodeling, the efficiency of synaptic transmission—as judged by the ratio of downstream to upstream electrical signals—remained stably poor but did not worsen with age.
Energy factories at the synapse under stress
To probe what happens before cells die, the authors compared the protein makeup of young mutant and normal retinas. The most striking early change was a broad decline in components of the tricarboxylic acid (TCA) cycle, a central pathway for generating energy. Microscopy confirmed that mitochondria, the organelles that host this cycle, were especially affected at photoreceptor synapses. In healthy mice, these terminals house prominent mitochondria; in α2δ4-deficient animals, the synaptic mitochondria were markedly smaller and fewer in number, particularly in cone terminals, long before widespread degeneration. Mitochondrial markers in the synaptic layer dropped, and some mitochondria appeared displaced into the layer of photoreceptor nuclei. By contrast, mitochondria in the inner segment—the main metabolic hub of the photoreceptor—were relatively preserved early on, highlighting a special vulnerability of synaptic mitochondria.
Broken calcium conversations between cell compartments
Because α2δ4 is part of a calcium channel complex, the team tested whether disrupted calcium handling might underlie the mitochondrial problems. In healthy photoreceptors, calcium entering through Cav1.4 channels at the synapse is closely coordinated with calcium storage and release by the endoplasmic reticulum (ER) and uptake by mitochondria, creating a microcircuit that links electrical activity to energy supply. In the mutant mice, proteins that release calcium from the ER (such as the ryanodine receptor RYR2) and a key mitochondrial calcium transporter (MCU) were specifically reduced in the synaptic layer. Markers of stress in the ER were elevated, suggesting that this compartment was also under strain. Together, these shifts point to a breakdown in the local calcium “crosstalk” among the cell membrane, ER, and mitochondria right where synaptic signaling occurs.
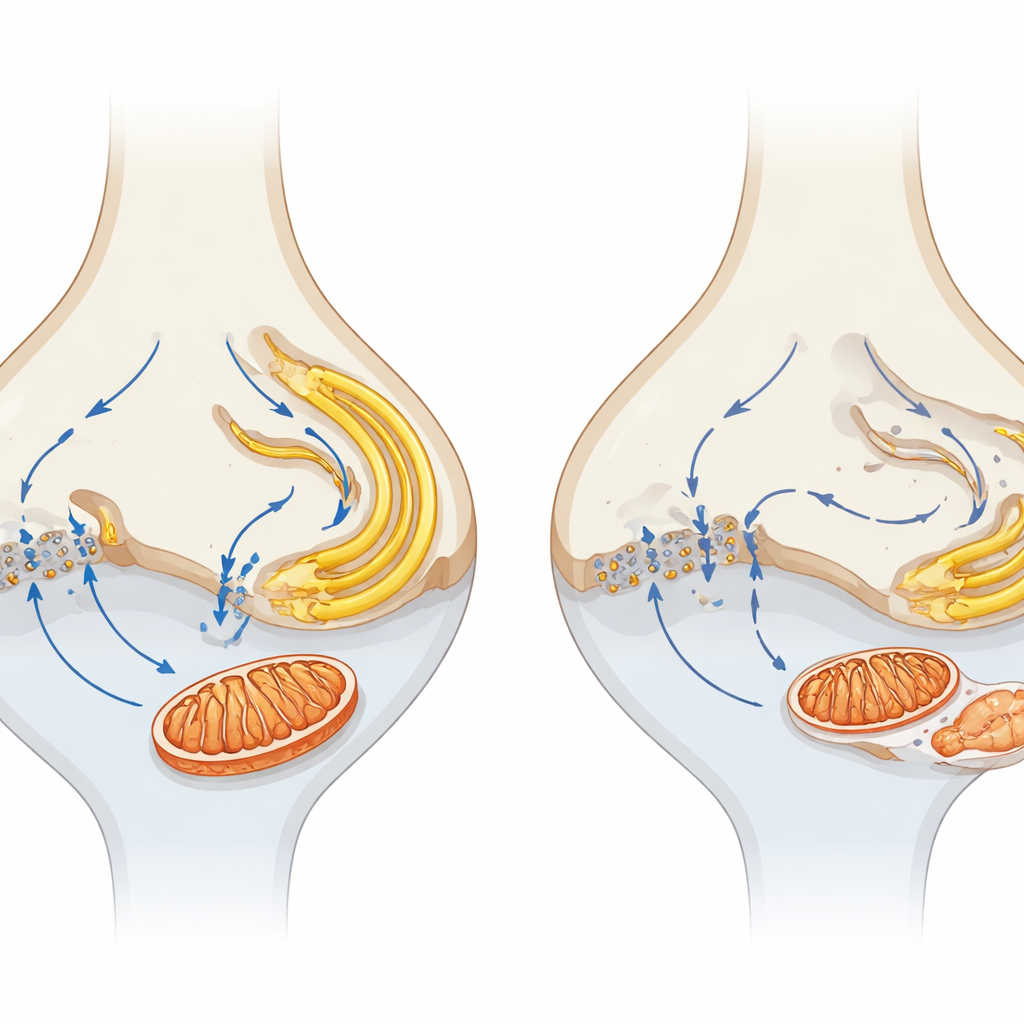
Connecting tiny synapses to long-term vision loss
By comparing α2δ4 knockout mice with animals completely lacking the Cav1.4 channel, the authors found that more severe channel disruption leads to faster and stronger degeneration, reinforcing the central role of calcium entry. They propose a model in which loss of α2δ4 weakens Cav1.4 function, disturbing calcium flow at the synapse and uncoupling it from nearby ER and mitochondria. Over time, this leads to damaged synaptic mitochondria, impaired energy production, and stress in internal membranes, which together prime photoreceptors for slow, progressive death. For a lay reader, the key insight is that healthy vision depends not just on having photoreceptors, but on keeping their tiny synaptic power plants and calcium signals finely tuned; targeting these early, local changes could offer new strategies to preserve sight in a broad range of retinal diseases.
Citation: Amieghemen, C.I., Ung, T.T., Huskin, G.N. et al. Dysfunction of α2δ4 leads to photoreceptor degeneration through disrupted synaptic mitochondria and calcium crosstalk. Cell Death Dis 17, 337 (2026). https://doi.org/10.1038/s41419-026-08587-3
Keywords: retinal degeneration, photoreceptors, mitochondria, calcium signaling, synaptic dysfunction