Clear Sky Science · de
Funktionsstörung von α2δ4 führt zu Photorezeptor-Degeneration durch gestörte synaptische Mitochondrien- und Kalzium‑Wechselwirkung
Warum diese Augenforschung wichtig ist
Sehverlust durch retinale Degeneration ist meist dauerhaft, und Therapien, die die lichtempfindlichen Zellen des Auges wirklich schützen, sind rar. Diese Studie stellt eine grundlegende Frage: Wie können kleine Probleme an den Verbindungen zwischen Zellen der Netzhaut allmählich zur Absterben dieser Zellen und schließlich zur Erblindung führen? Indem die Autorinnen und Autoren einem spezifischen genetischen Defekt nachgehen, der bei manchen Menschen mit erblichen Sehstörungen vorkommt, entlarven sie einen unerwarteten Übeltäter — kleine Energiefabriken an den Nervenenden der Photorezeptoren — und zeigen, wie gestörte Kalziumsignale an Synapsen nach und nach die Gesundheit der gesamten Zelle untergraben können.
Von fehlerhaftem Nachtsehen zu sterbenden Lichtwahrnehmern
Die Arbeit konzentriert sich auf Photorezeptoren, die Stäbchen und Zapfen, die Licht in elektrische Signale umwandeln. An ihrer Basis bilden diese Zellen spezialisierte Synapsen, die kontinuierlich chemische Signale an nachgeschaltete Neuronen freisetzen. Ein Protein namens α2δ4 hilft beim Zusammenbau und der Stabilisierung eines Kalziumkanal-Komplexes (Cav1.4) an diesen Synapsen, und Mutationen in seinem Gen wurden mit menschlichen retinalen Dystrophien in Verbindung gebracht. Patientinnen und Patienten zeigen häufig frühe Probleme bei der Signalübertragung von Photorezeptoren zu Bipolarzellen, verlieren aber möglicherweise über viele Jahre hinweg nicht großflächig Photorezeptoren. Mithilfe von Mäusen, denen α2δ4 fehlt, wollten die Forschenden verstehen, wie ein scheinbar „lokaler“ synaptischer Defekt schließlich zum langsamen Ausdünnen der Schicht mit den Photorezeptorzellkörpern führt.
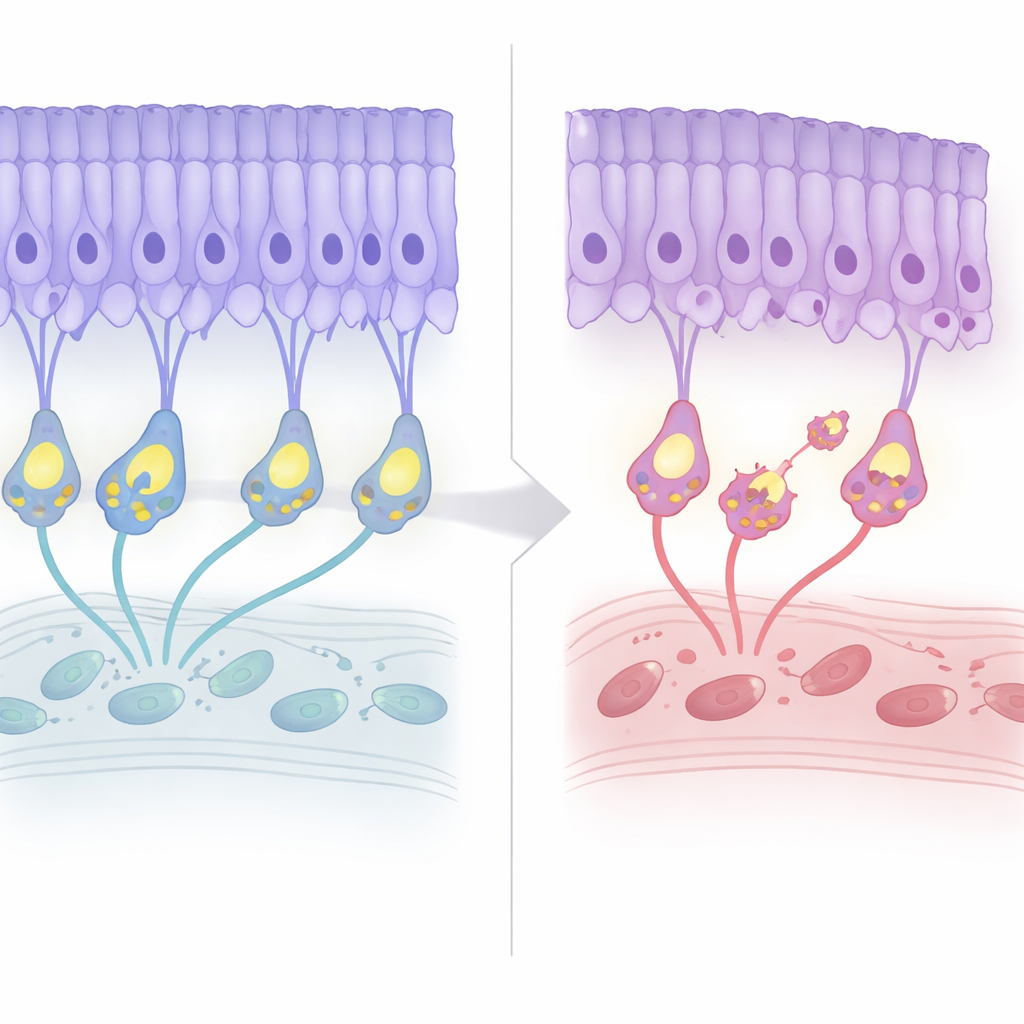
Langsamer struktureller Rückgang mit frühen Warnzeichen
Das Team verfolgte die Netzhäute dieser Mäuse über ihre Lebenszeit. In den ersten Monaten wirkte die Gesamtarchitektur weitgehend normal, und elektrische Messungen deuteten darauf hin, dass die Lichtwahrnehmung an sich größtenteils erhalten blieb. Etwa im Alter von sieben Monaten begann jedoch die Schicht der Photorezeptornuklei um etwa 20 Prozent auszudünnen und sich dann auf diesem Niveau einzupendeln, was die milde, spät einsetzende Degeneration widerspiegelt, die bei Patientinnen und Patienten beobachtet wird. Interessanterweise traten Marker des programmierten Zelltods früher auf, noch bevor ein offensichtlicher Zellverlust sichtbar war. Gleichzeitig reorganisierte sich die „Verdrahtung“: Photorezeptor-Terminals zogen sich aus ihrer üblichen Schicht zurück, und die Dendriten der Bipolarzellen wuchsen nach oben, um dort ektopische Verbindungen zu bilden. Trotz dieser Umstrukturierung blieb die Effizienz der synaptischen Übertragung — gemessen am Verhältnis der nachgeschalteten zu vorgelagerten elektrischen Signalen — dauerhaft gering, verschlechterte sich aber nicht mit dem Alter.
Synaptische Energiefabriken unter Stress
Um zu untersuchen, was vor dem Zelltod geschieht, verglichen die Autorinnen und Autoren die Proteinzusammensetzung junger Mutanten- und Normalnetzhäute. Die auffälligste frühe Veränderung war ein breiter Rückgang von Komponenten des Tricarbonsäure-(TCA-)Zyklus, eines zentralen Wegs zur Energiegewinnung. Die Mikroskopie bestätigte, dass Mitochondrien, die Organellen, die diesen Zyklus beherbergen, besonders an den Photorezeptor-Synapsen betroffen waren. Bei gesunden Mäusen findet man in diesen Terminals prominente Mitochondrien; bei α2δ4-defizienten Tieren waren die synaptischen Mitochondrien deutlich kleiner und weniger zahlreich, besonders in den Konusterminals, und dies lange bevor es zu weitverbreiteter Degeneration kam. Mitochondriale Marker in der synaptischen Schicht gingen zurück, und einige Mitochondrien schienen in die Schicht der Photorezeptornuklei verschoben zu sein. Im Gegensatz dazu blieben die Mitochondrien im inneren Segment — dem hauptsächlichen Stoffwechselzentrum des Photorezeptors — zunächst relativ erhalten, was eine besondere Verwundbarkeit der synaptischen Mitochondrien hervorhebt.
Gestörte Kalzium‑Kommunikation zwischen Zellkompartimenten
Da α2δ4 Teil eines Kalziumkanal-Komplexes ist, prüfte das Team, ob gestörte Kalziumhandhabung den mitochondrialen Problemen zugrunde liegen könnte. In gesunden Photorezeptoren ist das Kalzium, das über Cav1.4-Kanäle an der Synapse eintritt, eng mit der Kalziumspeicherung und -freisetzung im endoplasmatischen Retikulum (ER) und der Aufnahme durch Mitochondrien abgestimmt; so entsteht ein Mikrokreislauf, der elektrische Aktivität mit der Energieversorgung verknüpft. In den Mutantenmäusen waren Proteine, die Kalzium aus dem ER freisetzen (wie der Ryanodin-Rezeptor RYR2), und ein zentraler mitochondrialer Kalziumtransporter (MCU) spezifisch in der synaptischen Schicht vermindert. Marker für ER-Stress waren erhöht, was darauf hindeutet, dass auch dieses Kompartiment unter Belastung stand. Zusammengenommen deuten diese Veränderungen auf einen Zusammenbruch der lokalen Kalzium‑„Wechselgespräche“ zwischen Zellmembran, ER und Mitochondrien dort hin, wo synaptische Signalübertragung stattfindet.
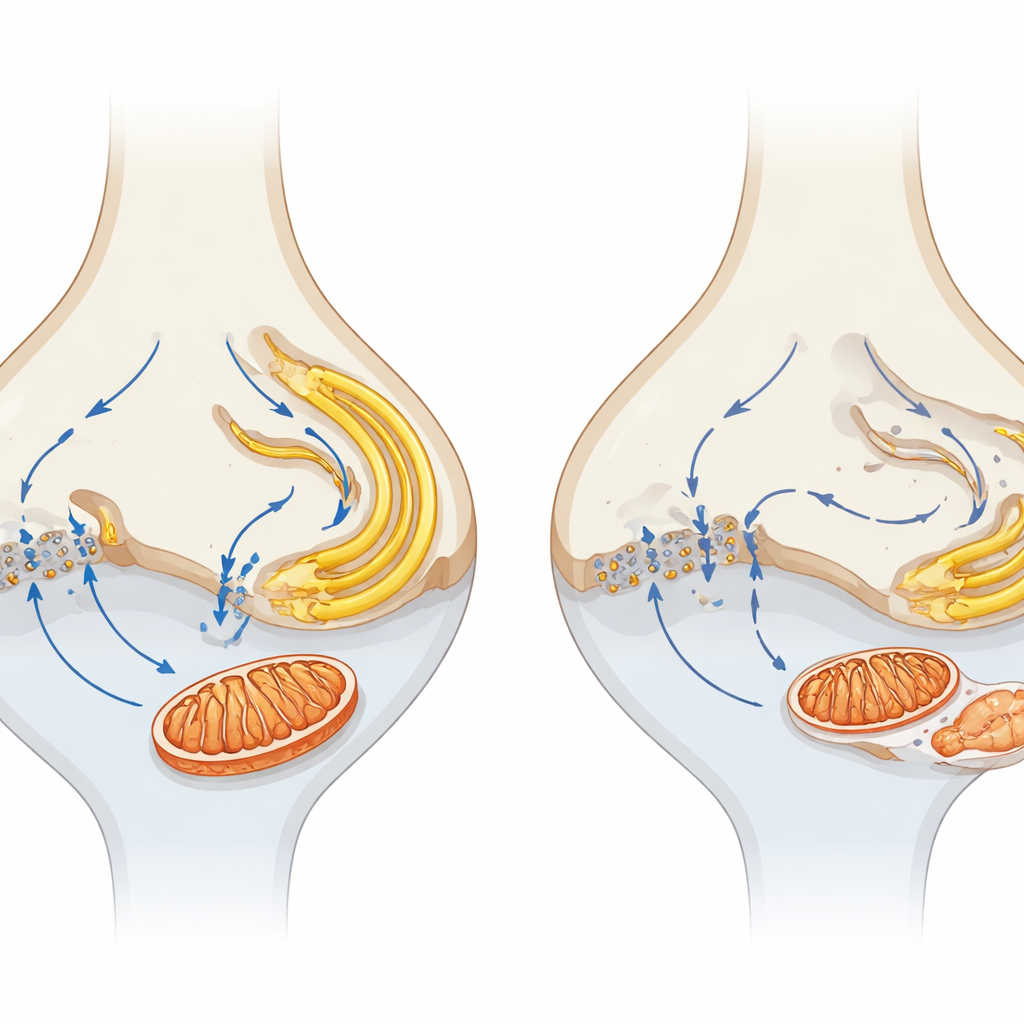
Kleine Synapsen — große Folgen für das langfristige Sehen
Durch den Vergleich von α2δ4-Knockout-Mäusen mit Tieren, denen der Cav1.4-Kanal vollständig fehlt, fanden die Forscherinnen und Forscher heraus, dass eine stärker ausgeprägte Kanalstörung zu schnellerer und intensiverer Degeneration führt, was die zentrale Rolle des Kalziumeinstroms unterstreicht. Sie schlagen ein Modell vor, in dem der Verlust von α2δ4 die Cav1.4-Funktion schwächt, den Kalziumfluss an der Synapse stört und die Kopplung zu nahegelegenen ER und Mitochondrien aufhebt. Im Laufe der Zeit führt dies zu beschädigten synaptischen Mitochondrien, beeinträchtigter Energieproduktion und Stress in internen Membranen, die zusammen die Photorezeptoren für einen langsamen, fortschreitenden Zelltod prädisponieren. Für eine nichtfachliche Leserschaft ist die zentrale Erkenntnis: Gesunde Sehkraft hängt nicht nur vom Vorhandensein von Photorezeptoren ab, sondern davon, dass ihre kleinen synaptischen Kraftwerke und Kalziumsignale fein abgestimmt bleiben; das gezielte Ansprechen dieser frühen, lokalen Veränderungen könnte neue Strategien bieten, um das Sehvermögen bei einer Vielzahl retinaler Erkrankungen zu erhalten.
Zitation: Amieghemen, C.I., Ung, T.T., Huskin, G.N. et al. Dysfunction of α2δ4 leads to photoreceptor degeneration through disrupted synaptic mitochondria and calcium crosstalk. Cell Death Dis 17, 337 (2026). https://doi.org/10.1038/s41419-026-08587-3
Schlüsselwörter: retinale Degeneration, Photorezeptoren, Mitochondrien, Kalziumsignalisierung, synaptische Dysfunktion