Clear Sky Science · en
Stochastic modeling of epigenetic memory
How Cells Remember Who They Are
Every cell in your body carries essentially the same DNA, yet a brain cell behaves very differently from a skin or immune cell. This lasting “sense of identity” must be copied each time cells divide, and when it fails, diseases like cancer can arise. This article explains how researchers use mathematical tools to understand epigenetic memory—the biochemical marks on DNA and its packaging that let cells remember long-term whether particular genes should stay on or off.
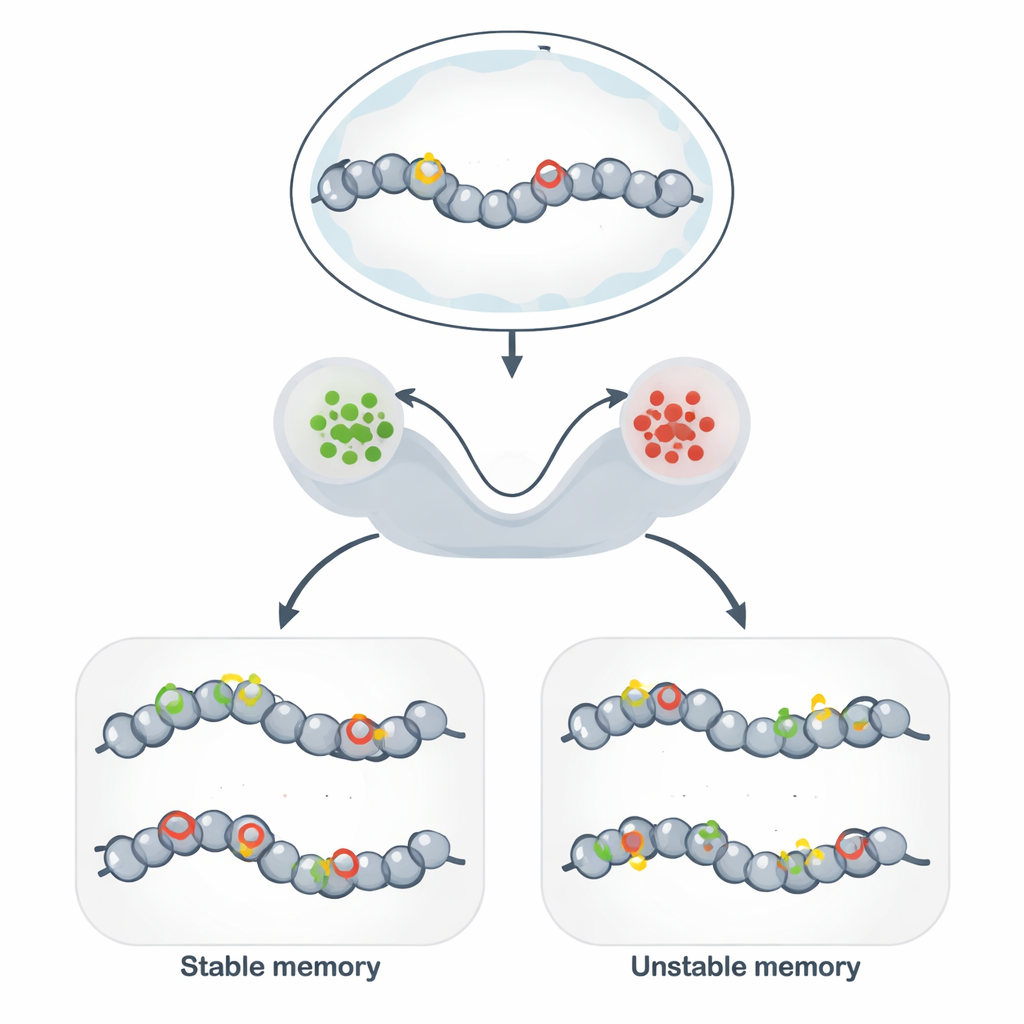
Beyond DNA: Stable Marks on the Genome
Epigenetic memory allows genetically identical cells to maintain different, stable behaviors over many cell divisions. Instead of changing DNA sequence, cells decorate DNA and its surrounding proteins with chemical tags that loosen or tighten the local structure. When tags favor an open structure, nearby genes tend to be active; when they favor a compact structure, genes are silenced. This mechanism underlies cell-type identity, the persistence of immune responses, some forms of brain memory, and promising tools for synthetic biology that aim to “program” stable cell states.
Why Randomness Cannot Be Ignored
At the molecular scale, the cell is a noisy place: key molecules often exist in small numbers and bump into each other at random. Traditional models that use smooth curves to describe how gene activity changes over time can predict multiple stable states, like two valleys separated by a hill. In such models, once the system rolls into a valley it should stay there forever. But in reality, random molecular events can occasionally kick the system over the hill into another valley. The article introduces stochastic (probabilistic) frameworks that treat reactions as random events and quantify how often such switches occur, for example through mean first-passage times that measure how long a state typically lasts before noise triggers a jump.
Mathematical Lenses on Molecular Chaos
The review describes two main ways of modeling this randomness. Stochastic chemical reaction networks track exact molecule counts and treat the system as a Markov process, where each reaction fires with a probability that depends on the present state. This approach can faithfully capture rare switches between stable gene-expression states but is computationally heavy. A second class of models uses stochastic differential equations, which approximate molecule counts as continuous quantities shaken by Gaussian noise. Variants such as the chemical Langevin equation and linear noise approximation trade some accuracy for analytical clarity, allowing researchers to derive how system parameters shape fluctuations around a stable state and how likely transitions between states become.
Chromatin Circuits as Memory Hardware
To connect these abstract models to biology, the authors focus on chromatin modification circuits: networks of chemical marks on histone proteins and DNA that reinforce one another. Histone tags that promote active chromatin, and those that promote repressed chromatin, can each recruit enzymes that spread the same tag to neighboring sites, creating powerful positive feedback. Stochastic models show that when this self-reinforcement is fast compared with erasure, the system develops two long-lived states—broadly “ON” and “OFF”—that function as binary memory. Adding DNA methylation, a highly stable tag copied during DNA replication, further biases the system toward long-lived repression and can dramatically extend how long a silenced state persists.
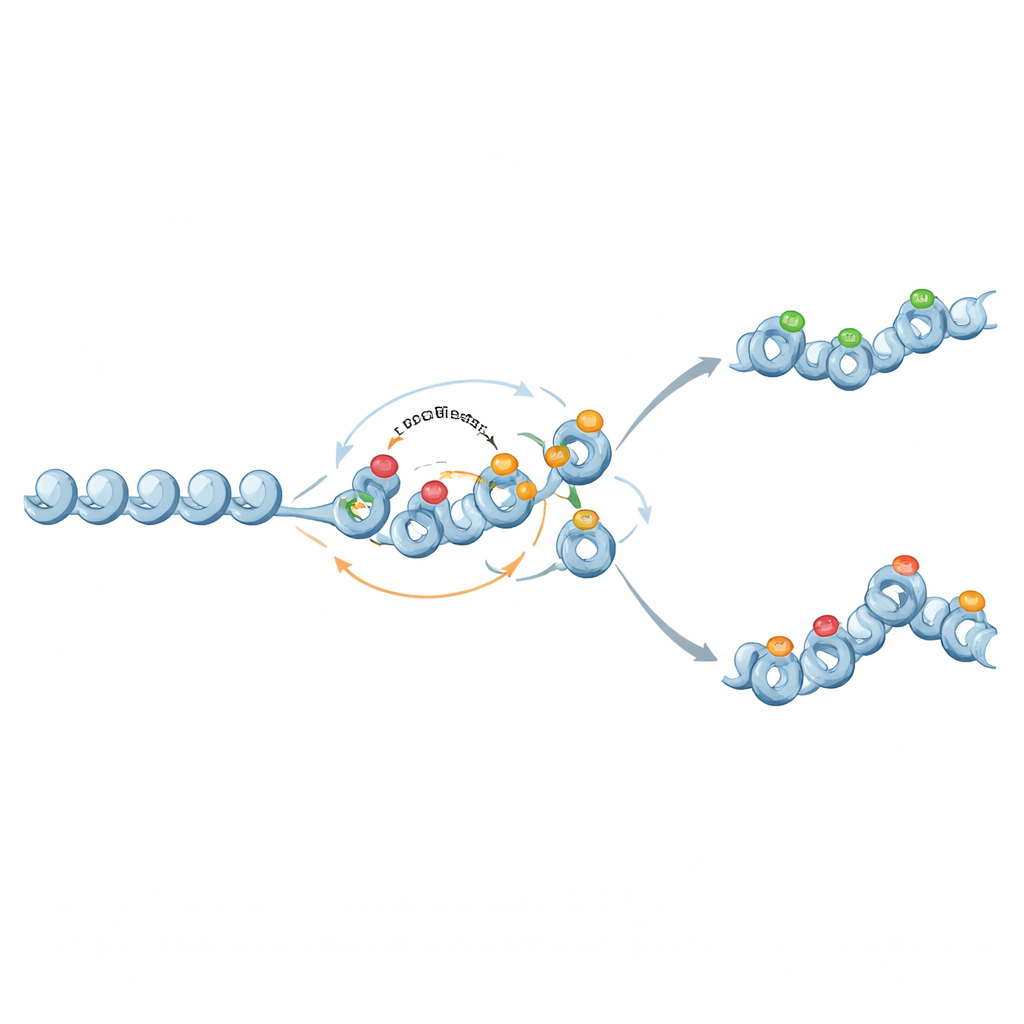
From Binary Switches to Graded Memories
Interestingly, the same molecular players can also support more nuanced, analog memory. When feedback from repressive histone marks to DNA methylation is weakened and methylation becomes nearly permanent but non-self-amplifying, methylation acts as a fixed backdrop that sets an average gene-expression level. Histone marks then fluctuate on top of this backdrop, sometimes producing two stable chromatin states and broad expression distributions at intermediate methylation levels. In this way, chromatin circuits can tune both how stable a gene’s activity is and how variable its expression can be across cells.
Why These Models Matter for Medicine and Design
The authors conclude that understanding epigenetic memory requires explicitly modeling molecular noise and the timing of key processes. Their synthesis highlights which reaction rates and feedback loops most strongly control the residence time of cells in active or repressed states. This insight can guide experiments that alter cell-cycle speed, enzyme levels, or specific chromatin marks to test model predictions. In the long run, such models may help engineer cells that can be reliably reprogrammed, hold a desired fate, or store information in a controllable way—opening possibilities for regenerative medicine, cancer therapy, and programmable immune responses.
Citation: Bruno, S., Del Vecchio, D. Stochastic modeling of epigenetic memory. npj Syst Biol Appl 12, 58 (2026). https://doi.org/10.1038/s41540-026-00664-9
Keywords: epigenetic memory, chromatin modifications, DNA methylation, stochastic modeling, gene regulation