Clear Sky Science · en
Molecular mechanisms of native ligand selectivity in catecholamine G protein-coupled receptors
How Cells Tell Similar Signals Apart
Our brain and body rely on tiny chemical messengers—like dopamine and adrenaline—to carry instructions between cells. These molecules look surprisingly alike, yet each must trigger the right response at the right place, whether it is sharpening attention, speeding the heart, or lifting mood. This study asks a deceptively simple question: how do almost identical molecules manage to “dial” different cellular phone numbers, and can we deliberately rewire those numbers for medicine and bioengineering?
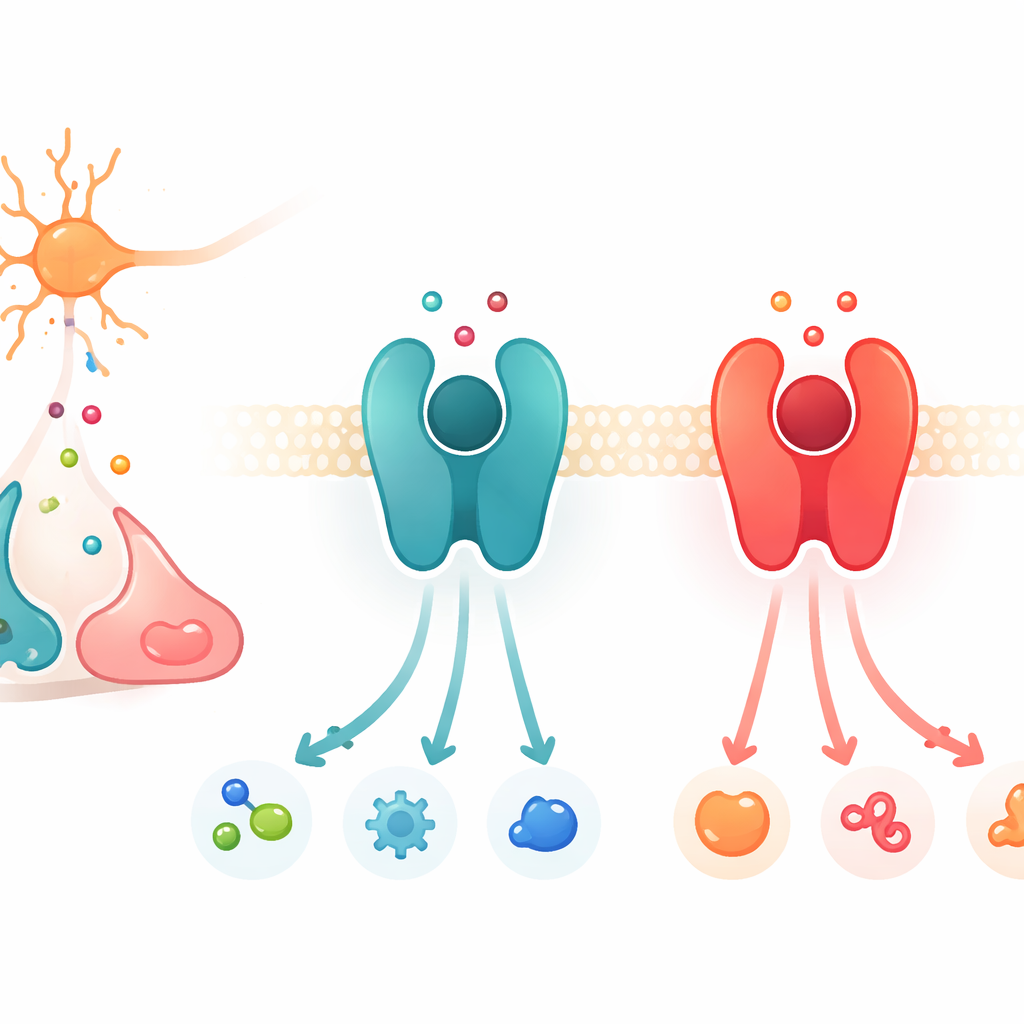
Signal Switchboards on Cell Surfaces
The work focuses on a large protein family called G protein–coupled receptors, or GPCRs. These proteins sit in the cell membrane and act like switchboards: when a chemical messenger binds outside, the receptor changes shape and activates partner proteins inside, launching a chain of signals. Among the many GPCRs, a subset responds to catecholamines, the closely related messengers dopamine, adrenaline, and noradrenaline. Two key examples are the β2-adrenergic receptor, which prefers adrenaline and noradrenaline, and the D1 dopamine receptor, which favors dopamine. Although these receptors and their binding pockets are strikingly similar in structure, each is highly picky about which messenger it responds to most strongly.
Swapping Preferences with Just a Few Changes
The researchers combined evolutionary comparisons, structural biology, and cell-based measurements to track down “hotspot” regions that determine this chemical preference. By aligning hundreds of receptor sequences from many species and scoring where the β2 and D1 families consistently differ, they found a short list of amino-acid positions that stood out. Some of these hotspots lie directly in the main binding pocket, where the messenger docks, while others sit slightly deeper in the protein, away from direct contact with the chemical. The team then systematically mutated these positions, testing more than a hundred receptor variants for how well they responded to dopamine, adrenaline, and noradrenaline using cellular readouts of signaling strength.
Tuning Pockets and Deep Control Knobs
Altering only the residues that touch the messenger did change selectivity, but often at a steep cost: the receptors became weak or almost unresponsive, even when they did start to favor a new ligand. The real breakthrough came when mutations in the binding pocket were combined with mutations in two deeper interlocking regions that act like internal control knobs. In the β2 receptor, adjustments in a tri-helix interface boosted its sensitivity and nudged it toward a dopamine-friendly mode without destroying function. In the D1 receptor, edits in a separate internal interface reorganized a network of side chains that indirectly reshape the pocket. With the right combinations, the researchers fully swapped the preferences: a modified β2 receptor now preferred dopamine, while a modified D1 receptor favored adrenaline and noradrenaline—yet both still signaled robustly.
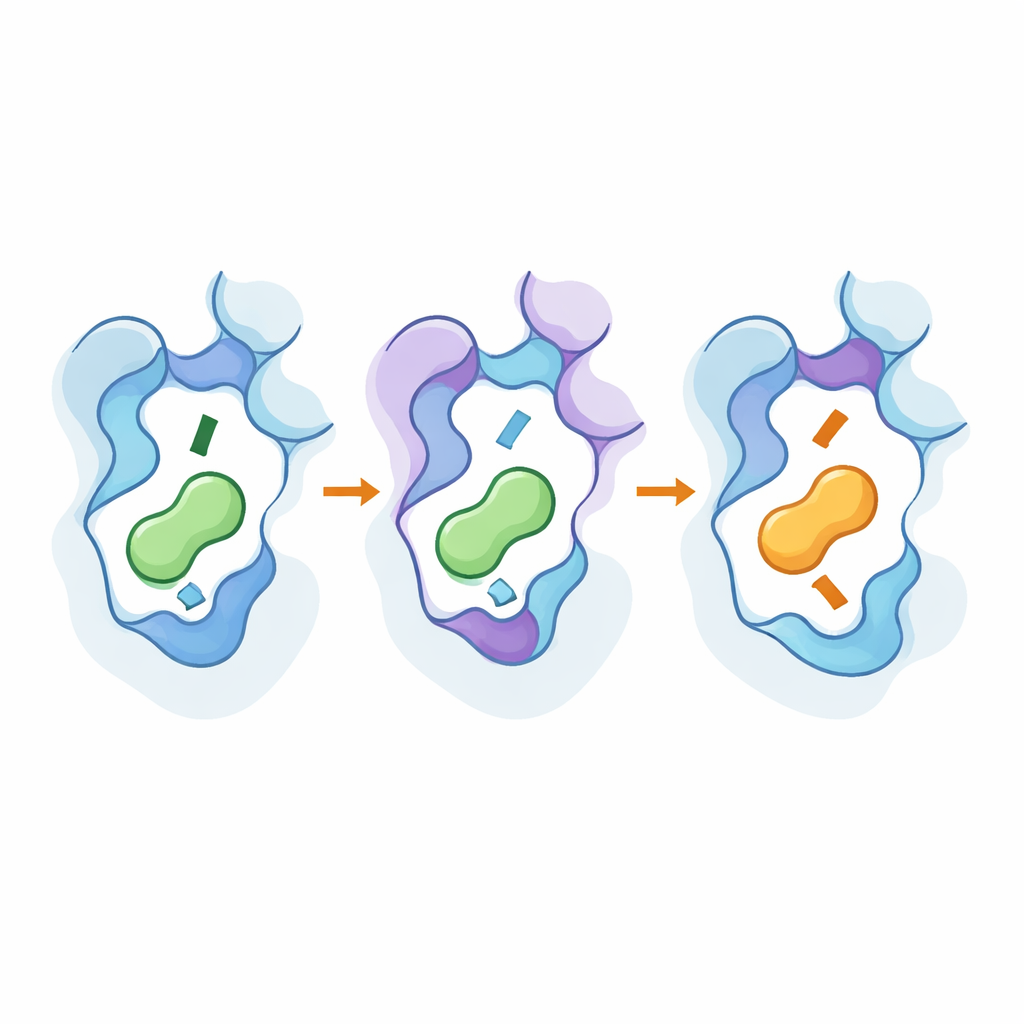
Peeking Under the Hood with High-Resolution Tools
To see how these changes worked at the atomistic level, the team used cryo-electron microscopy to solve 3D structures of the engineered receptors bound to their new preferred messengers, and ran extensive molecular dynamics simulations. The structures showed that key mutations subtly shifted parts of the receptor’s seventh helix and a conserved “toggle” residue, deepening or shallowing the pocket and changing how the amine end of the messenger sits. The distant hotspots helped stabilize active-like shapes and strengthened pathways that transmit the binding event to the inside of the protein. Simulations confirmed that networks of interacting residues link these outer and inner regions, and that the same small set of positions can be reused across related receptor subtypes to flip selectivity without making the receptors broadly promiscuous.
What This Means for Medicine and Evolution
In plain terms, the study shows that a handful of well-chosen “atoms” in these receptors act like tuning screws that decide whether a protein listens mainly to dopamine or to adrenaline. These screws are not all at the surface where the messenger docks; some are buried deeper, stabilizing how the whole structure flexes when a signal arrives. Because the same design logic recurs across receptor families, evolution can repeatedly repurpose similar protein scaffolds for new messengers by first boosting responsiveness via internal tweaks, then fine-tuning the pocket. For drug discovery, these insights point to precise spots—both in and around the binding site—that chemists can target to craft medicines that hit one receptor subtype while sparing others, and to engineer custom receptors that respond only to designed signals in therapies and biosensors.
Citation: Kahlous, N.A., Rinne, M.K., Zhang, X. et al. Molecular mechanisms of native ligand selectivity in catecholamine G protein-coupled receptors. Nat Commun 17, 4112 (2026). https://doi.org/10.1038/s41467-026-71361-8
Keywords: G protein-coupled receptors, dopamine, adrenaline, ligand selectivity, drug design