Clear Sky Science · de
Molekulare Mechanismen der Selektivität nativer Liganden in Katecholamin-G-Protein-gekoppelten Rezeptoren
Wie Zellen ähnliche Signale auseinanderhalten
Gehirn und Körper sind auf winzige chemische Botenstoffe angewiesen – etwa Dopamin und Adrenalin –, die Anweisungen zwischen Zellen übermitteln. Diese Moleküle sehen überraschend ähnlich aus, müssen aber dennoch jeweils die richtige Antwort am richtigen Ort auslösen, sei es zur Schärfung der Aufmerksamkeit, zur Beschleunigung des Herzens oder zur Stimmungsaufhellung. Die Studie stellt eine scheinbar einfache Frage: Wie schaffen es fast identische Moleküle, unterschiedliche „Telefonnummern“ der Zelle anzuwählen, und lässt sich diese Zuordnung gezielt für Medizin und Bioengineering umprogrammieren?
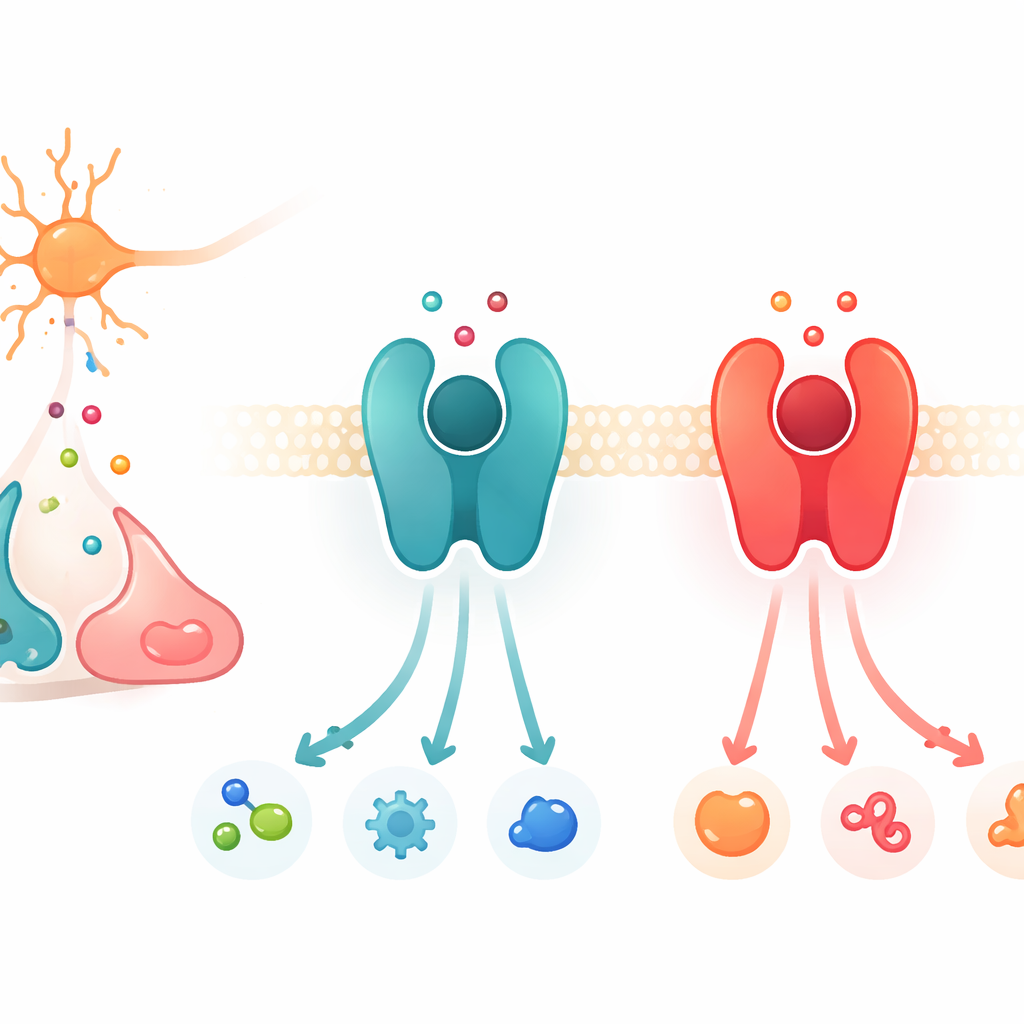
Signalvermittler an der Zelloberfläche
Die Arbeit konzentriert sich auf eine große Proteinfamilie, die G-Protein-gekoppelten Rezeptoren, kurz GPCRs. Diese Proteine sitzen in der Zellmembran und funktionieren wie Vermittlungsstellen: Bindet ein Botenstoff außen, ändert der Rezeptor seine Form und aktiviert Partnerproteine im Inneren, wodurch eine Signalkaskade ausgelöst wird. Unter den vielen GPCRs reagiert eine Untergruppe auf Katecholamine, die eng verwandten Botenstoffe Dopamin, Adrenalin und Noradrenalin. Zwei zentrale Beispiele sind der β2-adrenerge Rezeptor, der Adrenalin und Noradrenalin bevorzugt, und der D1-Dopaminrezeptor, der Dopamin favorisiert. Obwohl diese Rezeptoren und ihre Bindungstaschen strukturell auffallend ähnlich sind, ist jeder sehr wählerisch in Bezug auf den Botenstoff, auf den er am stärksten anspricht.
Präferenzen mit nur wenigen Änderungen tauschen
Die Forscher kombinierten evolutionäre Vergleiche, Strukturbiologie und zellbasierte Messungen, um „Hotspot“-Regionen zu identifizieren, die diese chemische Präferenz bestimmen. Durch das Ausrichten von Hunderten Rezeptorsequenzen aus vielen Arten und das Bewerten, wo sich die β2- und D1-Familien konstant unterscheiden, fanden sie eine kurze Liste von Aminosäurepositionen, die herausstachen. Einige dieser Hotspots liegen direkt in der Hauptbindetasche, wo der Botenstoff andockt, andere sitzen etwas tiefer im Protein, fern vom direkten Kontakt mit dem Molekül. Das Team mutierte diese Positionen systematisch und testete mehr als hundert Rezeptorvarianten darauf, wie gut sie auf Dopamin, Adrenalin und Noradrenalin reagierten, mithilfe zellulärer Messgrößen der Signalstärke.
Feinabstimmung der Tasche und tiefe Kontrollknöpfe
Allein die Veränderung der Reste, die den Botenstoff berühren, veränderte zwar die Selektivität, führte aber oft zu hohen Kosten: Die Rezeptoren wurden schwach oder fast nicht mehr ansprechbar, selbst wenn sie begannen, einen neuen Liganden zu bevorzugen. Der eigentliche Durchbruch gelang, als Mutationen in der Bindetasche mit Veränderungen in zwei tieferen, ineinandergreifenden Regionen kombiniert wurden, die wie interne Kontrollelemente wirken. Im β2-Rezeptor steigerten Anpassungen in einer Drei-Helix-Schnittstelle die Empfindlichkeit und schoben ihn in Richtung eines dopaminfreundlichen Modus, ohne die Funktion zu zerstören. Im D1-Rezeptor reorganisierten Änderungen in einer separaten internen Schnittstelle ein Netzwerk von Seitenketten, das die Tasche indirekt umformt. Mit den richtigen Kombinationen tauschten die Forscher die Präferenzen vollständig: Ein veränderter β2-Rezeptor bevorzugte nun Dopamin, während ein modifizierter D1-Rezeptor Adrenalin und Noradrenalin bevorzugte – und dennoch signalisierten beide weiterhin robust.
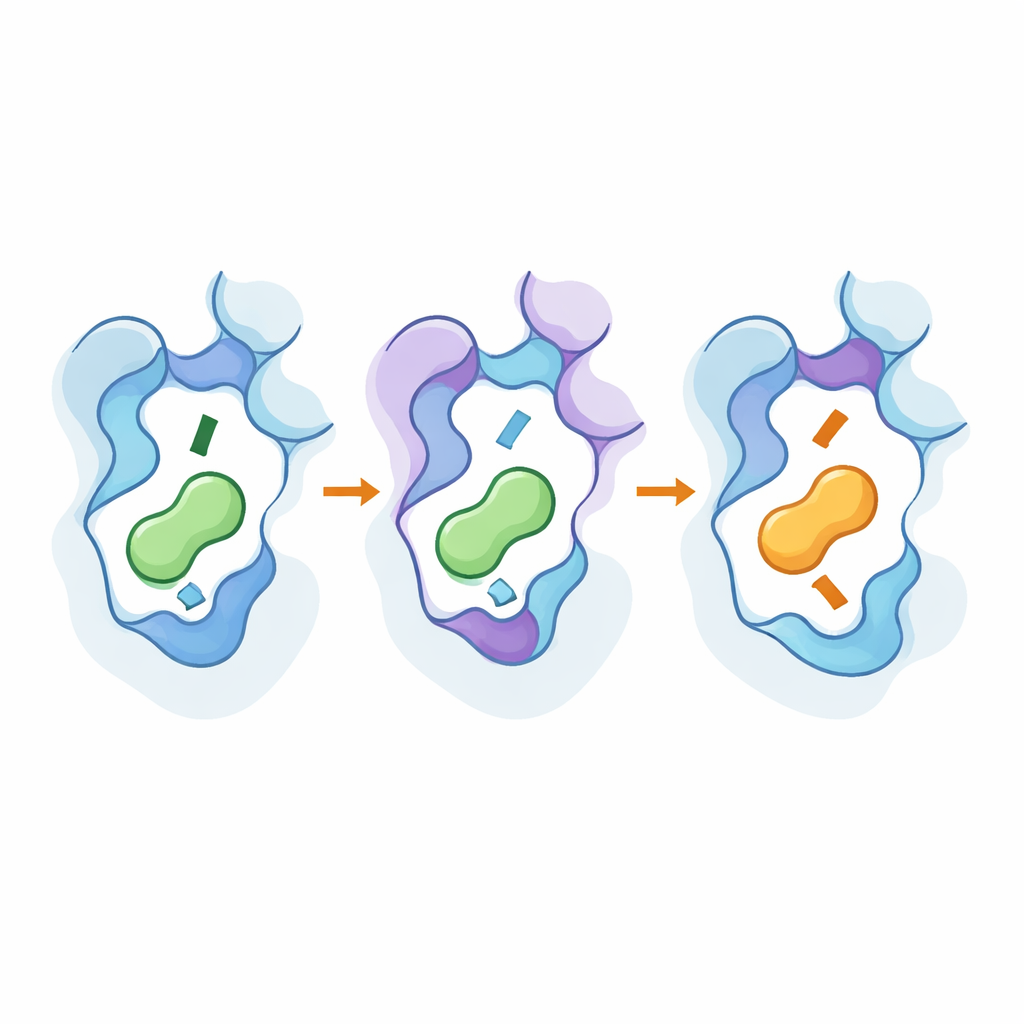
Ein Blick unter die Haube mit hochauflösenden Methoden
Um zu sehen, wie diese Änderungen auf atomarer Ebene funktionierten, verwendete das Team Kryo-Elektronenmikroskopie, um 3D-Strukturen der konstruierten Rezeptoren in Komplex mit ihren neuen bevorzugten Botenstoffen zu lösen, und führte umfangreiche Molekulardynamik-Simulationen durch. Die Strukturen zeigten, dass Schlüsselmutationen Teile der siebten Helix und eine konservierte „Toggle“-Restseite subtil verschoben, wodurch die Tasche sich vertiefte oder flacher wurde und sich die Lage des aminischen Endes des Botenstoffs veränderte. Die entfernten Hotspots halfen, aktiveähnliche Konformationen zu stabilisieren und verstärkten Pfade, die das Bindungsereignis nach innen übertragen. Simulationen bestätigten, dass Netzwerke wechselwirkender Reste diese äußeren und inneren Bereiche verbinden und dass dieselbe kleine Positionsmenge über verwandte Rezeptoruntertypen hinweg wiederverwendet werden kann, um Selektivität zu kippen, ohne die Rezeptoren allgemein promis-k zu machen.
Was das für Medizin und Evolution bedeutet
Einfach ausgedrückt zeigt die Studie, dass eine Handvoll wohlgewählter „Atome“ in diesen Rezeptoren wie Einstellschrauben wirken, die entscheiden, ob ein Protein hauptsächlich auf Dopamin oder auf Adrenalin hört. Diese Schrauben sitzen nicht alle an der Oberfläche, wo der Botenstoff andockt; einige sind tiefer vergraben und stabilisieren, wie die gesamte Struktur nachgibt, wenn ein Signal ankommt. Da dieselbe Designlogik in verschiedenen Rezeptorfamilien wiederkehrt, kann die Evolution ähnliche Proteingerüste wiederholt für neue Botenstoffe umnutzen, indem sie zunächst die Ansprechbarkeit durch interne Anpassungen erhöht und dann die Tasche feinjustiert. Für die Wirkstoffforschung deuten diese Einsichten auf präzise Stellen – sowohl in als auch um die Bindungsstelle herum –, die Chemiker anvisieren können, um Medikamente zu entwickeln, die einen Rezeptoruntertyp treffen und andere verschonen, und um maßgeschneiderte Rezeptoren zu entwerfen, die in Therapien und Biosensoren nur auf gezielt designte Signale reagieren.
Zitation: Kahlous, N.A., Rinne, M.K., Zhang, X. et al. Molecular mechanisms of native ligand selectivity in catecholamine G protein-coupled receptors. Nat Commun 17, 4112 (2026). https://doi.org/10.1038/s41467-026-71361-8
Schlüsselwörter: G-Protein-gekoppelte Rezeptoren, Dopamin, Adrenalin, Ligandenselektivität, Wirkstoffdesign