Clear Sky Science · zh
通过增量训练语言模型对药物分子进行结构优化
教会计算机改良药物分子
现代药物常常起源于有潜力但不完善的分子,化学家必须细致调整这些分子,才能将其发展为安全且有效的药物。本研究展示了一种将化学式“阅读”为语言的人工智能系统如何学会自行进行部分改良,提出新的候选药物,这些候选在效力上甚至超过已知最佳实例——且不依赖外部评分工具或基于试错的大量猜测。
为何优化药物分子如此困难
一旦研究人员找到能作用于生物靶点的初始分子,真正的工作才开始:将该早期“命中”分子转变为既强效又具选择性且适合用作药物的化合物。传统上,化学家会设计数十到数百个与原始结构相近的衍生物,在实验室合成并逐一测试。这些设计—合成—测试的循环需要多年的专业经验和大量实验投入。计算方法虽有帮助,但很多仅关注简单性质,例如分子的疏水性,而非其在生物学上产生的全面效应。还有些方法依赖独立的预测工具(“神谕”),用于估计活性,但这些工具对许多靶点可能不可靠或不可用。
用化学句子来指导设计
作者基于化学语言模型,这是一类将分子视为字符串(SMILES)并学习使结构在化学上合理且具生物学意义的“语法”和模式的深度学习系统。首先,他们在数十万已知生物活性分子上进行预训练,同时刻意过滤掉与后续研究特定靶点相关的任何内容。由此得到的通用模型理解化学,但对所选受体没有先验知识,确保后续取得的任何成功确实来自新训练而非起始数据中的隐性偏差。
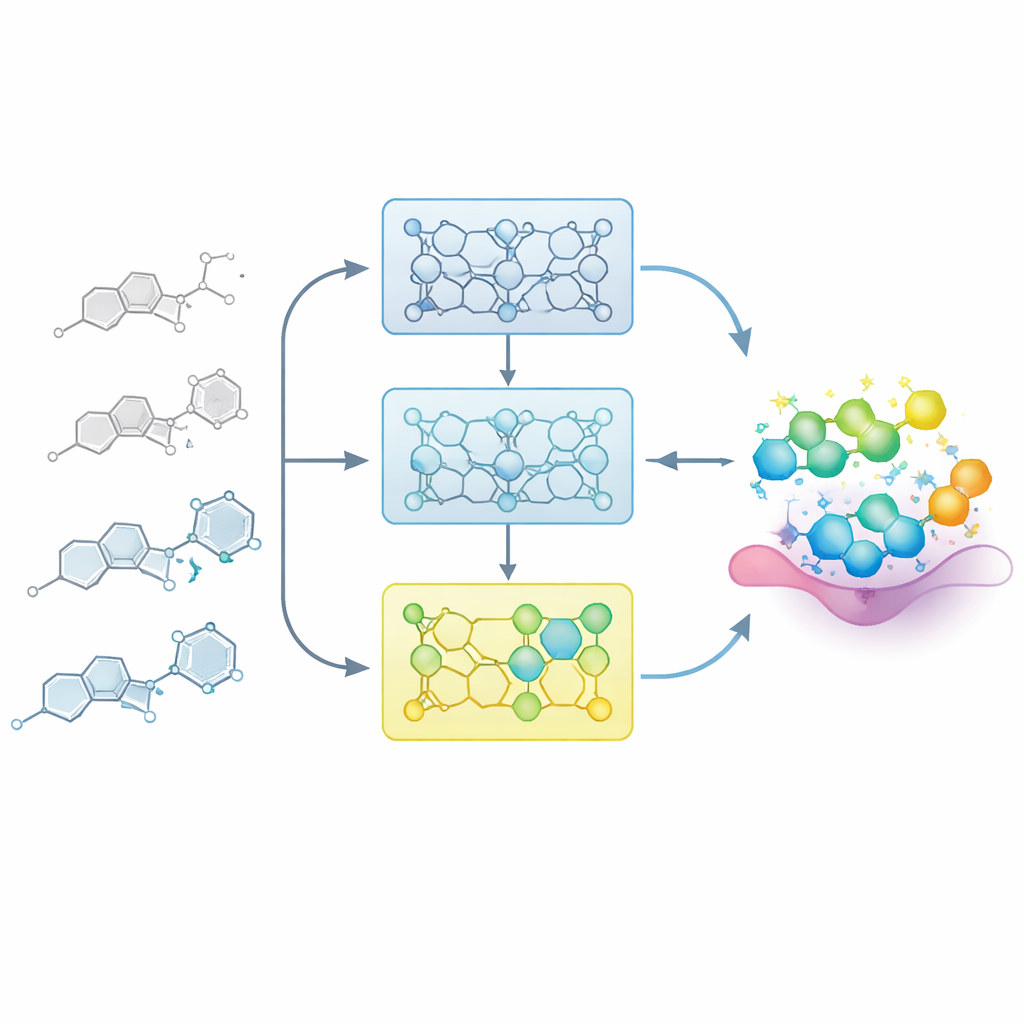
让模型像药物化学家那样学习
在真实的药物项目中,化学家会逐步建立结构与活性之间的映射:对核心骨架的小改动可能使化合物变弱或变强。研究者通过向模型输入精心排序的一系列相关分子(称为构效关系,SAR 系列)来模拟这一过程。他们不是一次性用所有已知样本来微调模型,而是将每个系列按效力划分为从弱到强的步骤。模型先接触活性较低的化合物,然后依次用包含更高效力分子的子集进行微调。这种“增量训练”创造了一条学习轨迹,将模型温和地引导到包含最佳分子的化学空间区域。
从理论到更强的候选药物
为检验这种训练策略是否真正有效,团队首先检查模型能否“重新发现”那些故意从训练中剔除的高活性分子。采用增量训练时,模型生成的排名靠前的设计与这些隐藏的高效化合物匹配的频率远高于一次性训练的模型,表明其已内化驱动高活性的模式。随后,作者针对两个具有医学相关性的靶点进行实际设计:参与代谢与炎症的 PPARγ,以及与免疫调节有关的 RORγ。在对每个靶点的已知配体进行增量训练后,模型提出了所选骨架的新类似物。当其中若干化合物被合成并在实验室测试时,所有九个 PPARγ 设计均成为高效激动剂,许多远超先前的最佳分子;而一项新的 RORγ 设计在结构上有所不同,同时活性几乎达到了该系列中已知最强化合物的水平。
对未来药物开发的意义
本研究表明,类似语言的模型不仅能发明分子,还能改进现有骨架,使其性能超越已知最佳实例——且不依赖外部评分工具——这指向了一种新的药物化学工作方式。增量训练方法使模型能够吸收细微的构效规则及其远程依赖关系,并将这些规则扩展到未探索的领域。对非专业读者而言,关键结论是:人工智能现在能更像受过数字训练的化学家助理,而非随机想法生成器,提出有针对性且可测试的改进方案,从而有望加速从早期命中到优化药物的进程。
引用: Hörmann, T., Mayer, D., Lewandowski, M. et al. Structural optimization of drug molecules with incrementally trained language models. Nat Commun 17, 3456 (2026). https://doi.org/10.1038/s41467-026-71591-w
关键词: 化学语言模型, 从头药物设计, 构效关系, 生成性化学, 人工智能在药物化学中的应用