Clear Sky Science · ru
Структурная оптимизация лекарственных молекул с помощью поэтапно обучаемых языковых моделей
Обучая компьютеры модифицировать лекарства
Современные лекарства часто начинаются как перспективные, но несовершенные молекулы, которые химикам приходится тщательно дорабатывать, чтобы они стали безопасными и эффективными препаратами. В этом исследовании показано, как система искусственного интеллекта, «читающая» химические формулы как язык, может научиться выполнять часть такой доработки самостоятельно, предлагая новые кандидаты в лекарства, которые оказываются даже более мощными, чем лучшие известные образцы — без опоры на внешние инструменты оценки или трудоёмкие пробные эксперименты.
Почему оптимизация лекарственных молекул так сложна
Как только исследователи находят первоначальную молекулу, влияющую на биологическую мишень, начинается настоящая работа: превратить этот ранний «хит» во что‑то сильное, селективное и пригодное в качестве лекарства. Традиционно химики проектируют десятки или сотни близких родственников исходной структуры, синтезируют их в лаборатории и тестируют каждый образец. Эти циклы «проектирование–создание–тестирование» требуют многолетнего опыта и больших экспериментальных усилий. Вычислительные методы пытались помочь, но многие из них ориентируются на простые свойства, такие как липофильность, а не на весь биологический эффект. Другие методы полагаются на отдельные предикторы («оракулы»), оценивающие активность, которые могут быть ненадёжны или недоступны для многих мишеней.
Использование химических «предложений» для управления дизайном
Авторы опираются на химические языковые модели — класс глубинных систем, который рассматривает молекулы как последовательности символов (SMILES) и изучает «грамматику» и шаблоны, делающие структуру химически осмысленной и биологически интересной. Сначала они предварительно обучают модель на сотнях тысяч известных биоактивных молекул, при этом сознательно исключая любые соединения, связанные с конкретными мишенями, которые будут изучаться позже. Это даёт универсальную модель, понимающую химию, но не имеющую предварочных знаний о выбранных рецепторах, что гарантирует: последующие успехи действительно обусловлены новым обучением, а не скрытой предвзятостью исходных данных.
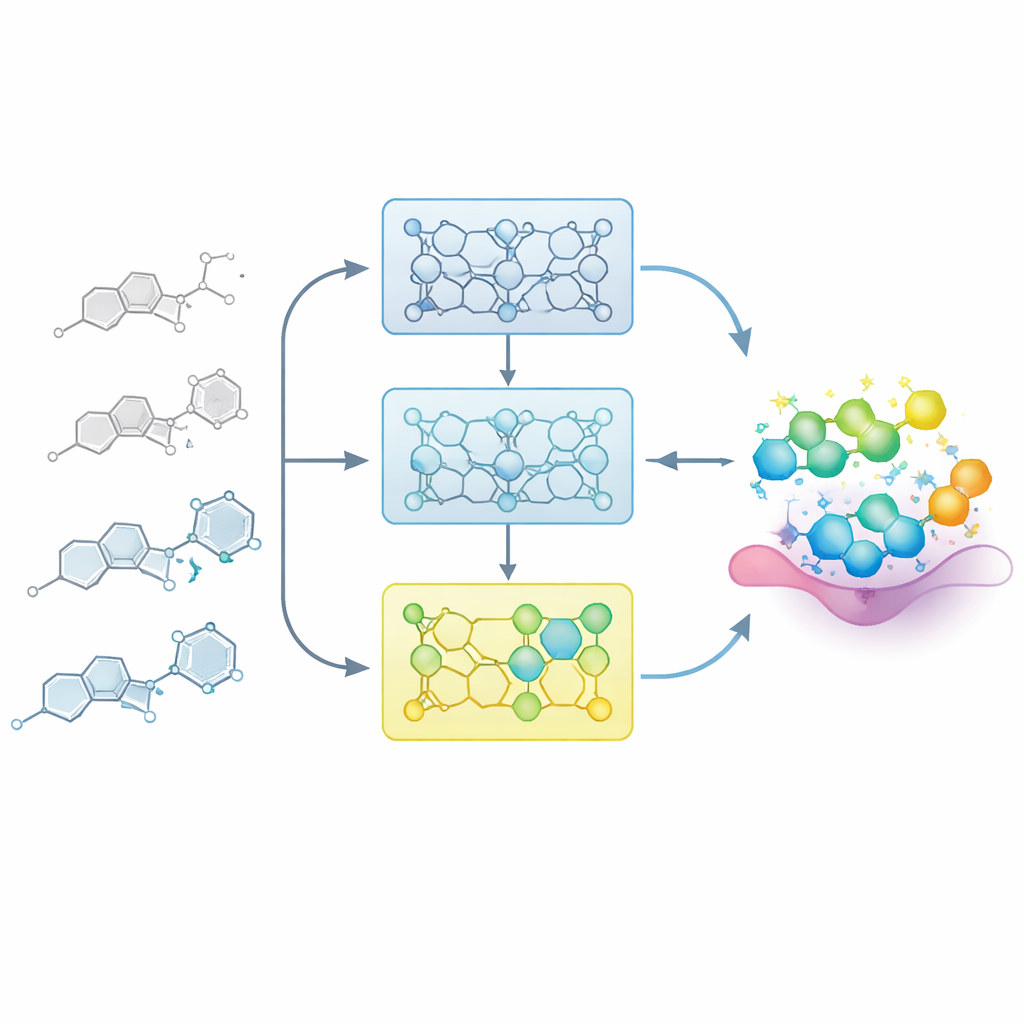
Позволяя модели учиться как медицинский химик
В реальных проектах по разработке лекарств химики постепенно выстраивают карту между структурой и активностью: небольшие изменения в ядре молекулы могут ослабить или усилить соединение. Исследователи имитируют этот процесс, подавая модели тщательно упорядоченные серии родственных молекул, называемые сериями структуро–активных зависимостей (SAR). Вместо того чтобы дообучать модель разом на всех известных примерах, они делят каждую серию на шаги по степени потенции — от менее активных к более активным членам. Модель сначала знакомят с менее активными соединениями, затем последовательно дообучают на поднаборы с более мощными примерами. Это «поэтапное обучение» создаёт траекторию усвоения, в которой модель мягко направляется в область химического пространства, где сосредоточены лучшие молекулы.
От теории к новым, более сильным кандидатам в лекарства
Чтобы проверить, действительно ли эта стратегия обучения помогает, команда сначала проверяет, может ли модель «вновь открыть» высокоактивные молекулы, которые целенаправленно исключили из обучения. При поэтапном обучении модель генерирует верхние по ранжиру дизайны, совпадающие с этими скрытыми мощными соединениями, гораздо чаще, чем модели, обученные за один шаг — это указывает на то, что она усвоила шаблоны, определяющие высокую активность. Авторы затем переходят к реальному дизайну для двух медицински значимых мишеней: PPARγ, вовлечённого в метаболизм и воспаление, и RORγ, связанного с регуляцией иммунитета. После поэтапного обучения на известных лигандах для каждой мишени модель предлагает новые аналоги выбранных скелетов. Когда несколько из этих соединений синтезировали и протестировали в лаборатории, все девять разработок для PPARγ оказались высокоактивными агонистами, многие значительно превзошли прежний лучший образец, а новый дизайн для RORγ почти достиг потенции самого сильного известного соединения в своей серии и при этом структурно отличался.
Что это означает для будущих лекарств
Показав, что модель в стиле языковой модели может не только придумывать молекулы, но и совершенствовать существующие скелеты так, чтобы они превосходили лучшие известные примеры — без опоры на внешние инструменты оценки — эта работа указывает на новый подход в медицинской химии. Подход поэтапного обучения позволяет модели усваивать тонкие правила структуравтивной активности и их дальнодействующие взаимосвязи, а затем распространять их в неизведанные области. Для неспециалистов главный вывод в том, что ИИ теперь может действовать не столько как генератор случайных идей, сколько как цифрово обученный помощник химика, предлагая целевые, тестируемые улучшения перспективных молекул и потенциально ускоряя путь от ранних хитов к оптимизированным препаратам.
Цитирование: Hörmann, T., Mayer, D., Lewandowski, M. et al. Structural optimization of drug molecules with incrementally trained language models. Nat Commun 17, 3456 (2026). https://doi.org/10.1038/s41467-026-71591-w
Ключевые слова: языковые модели химии, de novo дизайн лекарств, структуро–активные зависимости, генеративная химия, ИИ в медицинской химии