Clear Sky Science · fr
Optimisation structurelle des molécules médicamenteuses avec des modèles de langage entraînés de façon incrémentale
Apprendre aux ordinateurs à bricoler des médicaments
Les médicaments modernes commencent souvent par des molécules prometteuses mais imparfaites que les chimistes doivent ajuster minutieusement pour en faire des médicaments sûrs et efficaces. Cette étude montre comment un système d’intelligence artificielle qui « lit » les formules chimiques comme un langage peut apprendre à accomplir une partie de ces ajustements de manière autonome, en proposant de nouveaux candidats-médicaments encore plus puissants que les meilleurs exemplaires connus — sans s’appuyer sur des outils d’évaluation externes ni sur des tâtonnements fondés sur l’intuition.
Pourquoi l’optimisation des molécules médicamenteuses est si difficile
Une fois que les chercheurs trouvent une molécule initiale qui affecte une cible biologique, le vrai travail commence : transformer ce premier « hit » en quelque chose de puissant, sélectif et approprié comme médicament. Traditionnellement, les chimistes conçoivent des dizaines ou des centaines de proches analogues de la structure initiale, les synthétisent en laboratoire et testent chacun. Ces cycles conception–synthèse–test exigent des années d’expertise et des efforts expérimentaux considérables. Les méthodes informatiques ont tenté d’aider, mais beaucoup se concentrent sur des propriétés simples, comme l’hydrophobicité d’une molécule, plutôt que sur l’effet biologique global qu’elle produit. D’autres approches dépendent d’outils de prédiction séparés (« oracles ») qui estiment l’activité et peuvent être peu fiables ou indisponibles pour de nombreuses cibles.
Utiliser les phrases chimiques pour guider la conception
Les auteurs s’appuient sur des modèles de langage chimiques, un type de système d’apprentissage profond qui traite les molécules comme des chaînes de caractères (SMILES) et apprend la « grammaire » et les motifs qui rendent une structure chimiquement sensée et potentiellement active biologiquement. D’abord, ils préentraînent un modèle sur des centaines de milliers de molécules bioactives connues, en filtrant délibérément tout ce qui est lié aux cibles spécifiques qu’ils étudieront ensuite. Cela produit un modèle généraliste qui comprend la chimie mais n’a aucune connaissance préalable des récepteurs choisis, garantissant que tout succès ultérieur provienne véritablement du nouvel entraînement reçu plutôt que d’un biais caché dans les données de départ.
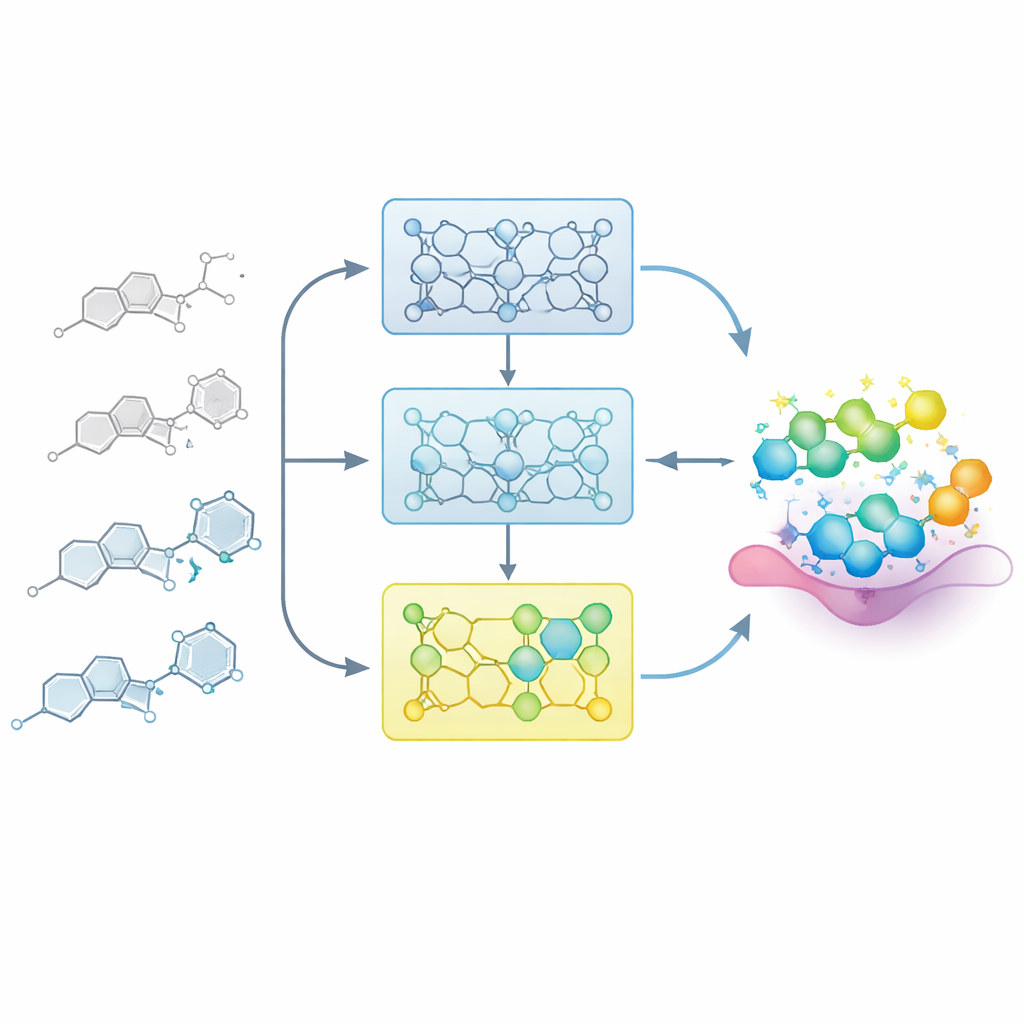
Laisser le modèle apprendre comme un chimiste médicinal
Dans de vrais projets pharmaceutiques, les chimistes construisent progressivement une carte entre structure et activité : de petits changements sur un squelette central peuvent affaiblir ou renforcer un composé. Les chercheurs reproduisent ce processus en alimentant le modèle avec des séries ordonnées de molécules apparentées, appelées séries de relations structure–activité (SAR). Plutôt que d’affiner le modèle en une seule fois sur tous les exemples connus, ils divisent chaque série en étapes basées sur la puissance, des membres les moins actifs aux plus actifs. Le modèle est d’abord exposé aux composés les moins actifs, puis affiné successivement avec des sous-ensembles contenant des exemples plus puissants. Cet « entraînement incrémental » crée une trajectoire d’apprentissage dans laquelle le modèle est guidé progressivement vers la région de l’espace chimique où résident les meilleures molécules.
De la théorie à de nouveaux candidats-médicaments plus puissants
Pour vérifier si cette stratégie d’entraînement aide réellement, l’équipe vérifie d’abord si le modèle peut « redécouvrir » des molécules fortement actives qui ont été délibérément exclues de l’entraînement. Avec l’entraînement incrémental, le modèle génère des conceptions bien classées qui correspondent à ces composés puissants cachés beaucoup plus souvent que les modèles entraînés en une seule étape, ce qui indique qu’il a intériorisé les motifs qui gouvernent une forte activité. Les auteurs passent ensuite à la conception en conditions réelles pour deux cibles d’intérêt médical : PPARγ, impliqué dans le métabolisme et l’inflammation, et RORγ, impliqué dans la régulation immunitaire. Après un entraînement incrémental sur les ligands connus de chaque cible, le modèle propose de nouveaux analogues de squelettes choisis. Lorsque plusieurs de ces composés sont synthétisés et testés en laboratoire, les neuf conceptions pour PPARγ se révèlent toutes être des agonistes très puissants, beaucoup dépassant la meilleure molécule précédente, et une nouvelle conception pour RORγ atteint presque la puissance du composé le plus fort connu dans sa série tout en étant structurellement distincte.
Ce que cela signifie pour les médicaments de demain
En montrant qu’un modèle de type langage peut non seulement inventer des molécules mais aussi affiner des squelettes existants pour surpasser les meilleurs exemplaires connus — sans s’appuyer sur des outils d’évaluation externes — ce travail ouvre une nouvelle façon de faire de la chimie médicinale. L’approche d’entraînement incrémental permet au modèle d’absorber des règles subtiles structure–activité et leurs interdépendances à longue portée, puis de les étendre vers des territoires inexplorés. Pour le grand public, l’idée principale est que l’IA peut désormais agir moins comme un générateur d’idées aléatoires et davantage comme l’assistant numérique d’un chimiste entraîné, proposant des améliorations ciblées et testables pour des molécules prometteuses et accélérant potentiellement le chemin des premiers hits vers des médicaments optimisés.
Citation: Hörmann, T., Mayer, D., Lewandowski, M. et al. Structural optimization of drug molecules with incrementally trained language models. Nat Commun 17, 3456 (2026). https://doi.org/10.1038/s41467-026-71591-w
Mots-clés: modèles de langage chimiques, conception de médicaments de novo, relations structure–activité, chimie générative, IA en chimie médicinale