Clear Sky Science · de
Strukturoptimierung von Arzneimittelmolekülen mit inkrementell trainierten Sprachmodellen
Computern beibringen, an Arzneimitteln zu tüfteln
Moderne Medikamente beginnen oft als vielversprechende, aber unvollkommene Moleküle, die Chemiker sorgfältig verändern müssen, damit sie sicher und wirksam werden. Diese Studie zeigt, wie ein System der künstlichen Intelligenz, das chemische Formeln wie eine Sprache „liest“, selbstständig einen Teil dieser Feinabstimmung erlernen kann und neue Wirkstoffkandidaten vorschlägt, die noch potenter sind als die bisher besten Beispiele — ohne auf externe Bewertungswerkzeuge oder raten- und trial‑and‑error‑lastige Verfahren angewiesen zu sein.
Warum die Optimierung von Arzneimittelmolekülen so schwierig ist
Sobald Forscher ein Ausgangsmolekül gefunden haben, das ein biologisches Ziel beeinflusst, beginnt die eigentliche Arbeit: aus diesem frühen „Hit“ etwas Starkes, Selektives und als Arzneimittel Geeignetes zu machen. Klassischerweise entwerfen Chemiker Dutzende bis Hunderte naher Verwandter der Ursprungsstruktur, synthetisieren sie im Labor und testen jedes einzelne. Diese Design‑Make‑Test‑Zyklen erfordern jahrelange Erfahrung und umfangreiche experimentelle Anstrengungen. Computergestützte Methoden haben versucht zu helfen, konzentrieren sich aber oft auf einfache Eigenschaften, etwa wie fettlöslich ein Molekül ist, statt auf die volle biologische Wirkung. Andere Ansätze verlassen sich auf getrennte Vorhersagewerkzeuge („Orakel“), die Aktivität schätzen — diese sind für viele Ziele unzuverlässig oder gar nicht verfügbar.
Chemische Sätze als Leitfaden für das Design
Die Autoren bauen auf chemischen Sprachmodellen auf, einer Form des Deep Learning, die Moleküle als Zeichenketten (SMILES) behandelt und die „Grammatik“ und Muster lernt, die eine Struktur chemisch sinnvoll und biologisch interessant machen. Zunächst trainieren sie ein Modell auf Hunderttausenden bekannter bioaktiver Moleküle, wobei bewusst alles entfernt wird, was mit den spezifischen Zielen zusammenhängt, die später untersucht werden. So entsteht ein generalistisches Modell, das Chemie versteht, aber keine Vorwissen über die gewählten Rezeptoren besitzt — das stellt sicher, dass spätere Erfolge wirklich aus dem neuen Training resultieren und nicht aus verborgenem Bias der Ausgangsdaten.
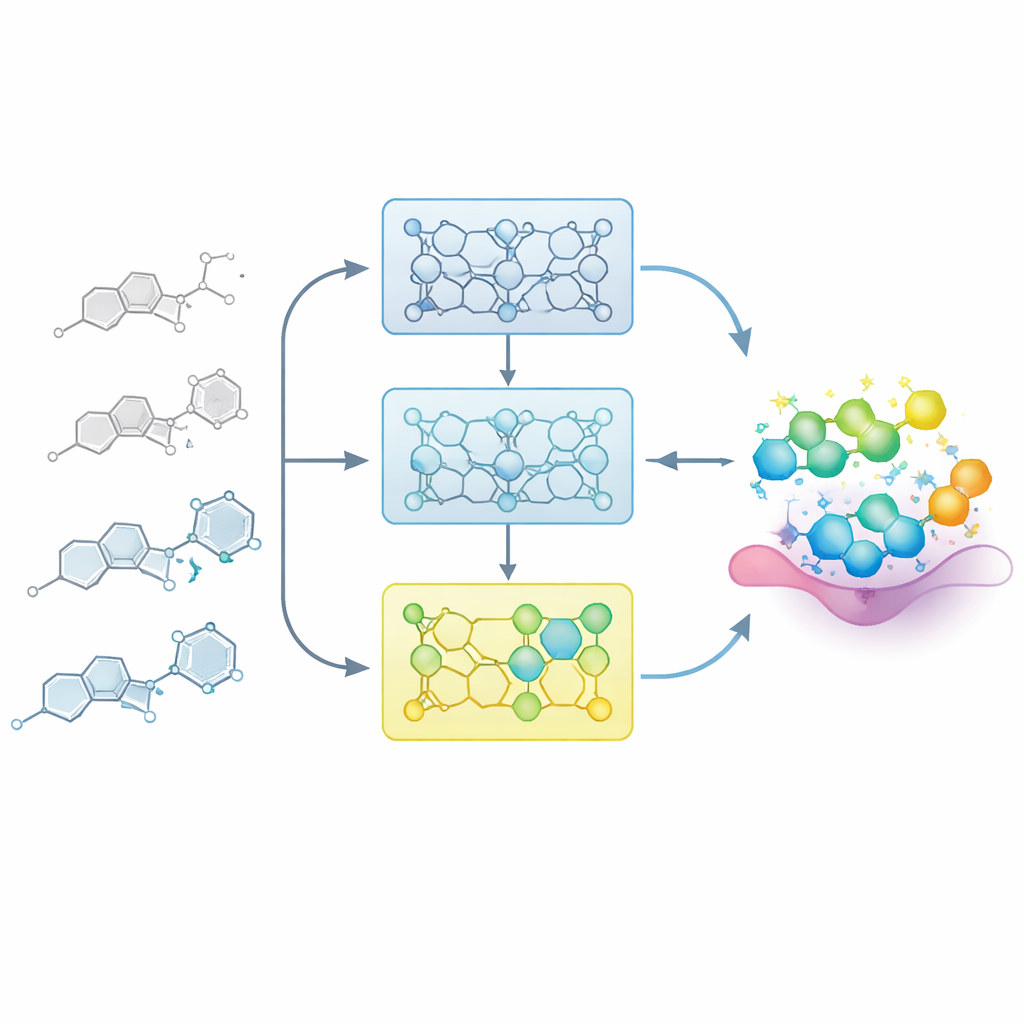
Das Modell wie einen medizinischen Chemiker lernen lassen
In realen Arzneimittelprojekten bauen Chemiker allmählich eine Landkarte zwischen Struktur und Aktivität auf: kleine Änderungen an einem Kerngerüst können eine Verbindung schwächen oder verstärken. Die Forschenden ahmen diesen Prozess nach, indem sie dem Modell sorgfältig geordnete Serien verwandter Moleküle zuführen, sogenannte Struktur‑Wirkungs‑Beziehungs (SAR)‑Serien. Statt das Modell in einem Schritt mit allen bekannten Beispielen zu verfeinern, teilen sie jede Serie in Stufen nach Potenz auf — von weniger zu stärker aktiven Mitgliedern. Das Modell wird zuerst mit den weniger aktiven Verbindungen vertraut gemacht und dann sukzessive mit Teilsätzen mit zunehmend potenteren Beispielen feinjustiert. Dieses „inkrementelle Training“ schafft eine Lernkurve, die das Modell behutsam in die Region des chemischen Raums führt, in der sich die besten Moleküle befinden.
Von der Theorie zu neuen, stärkeren Wirkstoffkandidaten
Um zu prüfen, ob diese Trainingsstrategie tatsächlich hilft, testen die Autoren zunächst, ob das Modell hochaktive Moleküle „wiederentdecken“ kann, die bewusst aus dem Training herausgehalten wurden. Mit inkrementellem Training erzeugt das Modell deutlich häufiger Top‑Entwürfe, die diesen versteckten potenten Verbindungen entsprechen, als Modelle, die in einem Schritt trainiert wurden — ein Hinweis darauf, dass es die Muster hoher Aktivität verinnerlicht hat. Danach wenden die Forschenden die Methode auf reale Designaufgaben für zwei medizinisch relevante Ziele an: PPARγ, beteiligt an Stoffwechsel und Entzündungen, und RORγ, das in der Immunregulation eine Rolle spielt. Nach inkrementellem Training mit bekannten Liganden für jedes Ziel schlägt das Modell neue Analoga bestimmter Gerüste vor. Als mehrere dieser Vorschläge synthetisiert und im Labor getestet werden, erweisen sich alle neun PPARγ‑Entwürfe als hochpotente Agonisten, viele übertreffen das bisher beste Molekül deutlich, und ein neuer RORγ‑Entwurf erreicht nahezu die Potenz der stärksten bekannten Verbindung seiner Serie, während er strukturell unterscheidbar bleibt.
Was das für zukünftige Medikamente bedeutet
Indem gezeigt wird, dass ein sprachähnliches Modell nicht nur Moleküle erfinden, sondern bestehende Gerüste so verfeinern kann, dass sie die besten bekannten Beispiele übertreffen — ohne sich auf externe Bewertungswerkzeuge zu stützen —, weist diese Arbeit auf einen neuen Weg in der medizinischen Chemie hin. Der inkrementelle Trainingsansatz ermöglicht es dem Modell, subtile Struktur‑Wirkungs‑Regeln und deren weitreichende Wechselwirkungen zu erfassen und diese dann in bislang unerforschte Bereiche auszudehnen. Für Nicht‑Spezialisten lautet die Kernbotschaft: KI kann jetzt weniger wie ein Zufallsgenerator und mehr wie die digital geschulte Assistentin eines Chemikers agieren, fokussierte, testbare Verbesserungen an vielversprechenden Wirkstoffmolekülen vorschlagen und so den Weg von ersten Hits zu optimierten Arzneimitteln potenziell beschleunigen.
Zitation: Hörmann, T., Mayer, D., Lewandowski, M. et al. Structural optimization of drug molecules with incrementally trained language models. Nat Commun 17, 3456 (2026). https://doi.org/10.1038/s41467-026-71591-w
Schlüsselwörter: chemische Sprachmodelle, de-novo-Arzneimittel-Design, Struktur-Wirkungs-Beziehungen, generative Chemie, KI in der medizinischen Chemie