Clear Sky Science · sv
Strukturoptimering av läkemedelsmolekyler med inkrementellt tränade språkmodeller
Lära datorer att mecka med läkemedel
Moderna läkemedel börjar ofta som lovande men ofullkomliga molekyler som kemister måste justera omsorgsfullt för att bli säkra och kraftfulla läkemedel. Denna studie visar hur ett artificiellt intelligenssystem som ”läser” kemiska formler som ett språk kan lära sig utföra en del av detta justerande på egen hand och föreslå nya läkemedelskandidater som är ännu mer potenta än de bästa kända exemplen — utan att förlita sig på externa poängsättningsverktyg eller tidsödande gissningsbaserade försök och fel.
Varför det är så svårt att optimera läkemedelsmolekyler
När forskare hittar en initial molekyl som påverkar ett biologiskt mål börjar det verkliga arbetet: att omvandla den tidiga ”träffen” till något som är potent, selektivt och lämpligt som medicin. Traditionellt designar kemister dussintals eller hundratals närbesläktade varianter av den ursprungliga strukturen, syntetiserar dem i labbet och testar varje enskild. Dessa design–make–test-cykler kräver år av expertis och omfattande experimentella insatser. Datorbaserade metoder har försökt hjälpa till, men många fokuserar på enkla egenskaper, såsom hur fet en molekyl är, snarare än den fulla biologiska påverkan den ger. Andra metoder är beroende av separata förutsägelseverktyg (”orakler”) som uppskattar aktivitet och som kan vara opålitliga eller otillgängliga för många mål.
Använda kemiska meningar för att styra designen
Författarna bygger vidare på kemiska språkmodeller, en typ av djupinlärningssystem som behandlar molekyler som strängar av tecken (SMILES) och lär sig den ”grammatik” och de mönster som gör en struktur kemiskt rimlig och biologiskt intressant. Först förtränar de en modell på hundratusentals kända bioaktiva molekyler, samtidigt som de avsiktligt filtrerar bort allt som är relaterat till de specifika målen de senare studerar. Detta ger en generalistmodell som förstår kemi men saknar förhandskunskap om de valda receptorerna, vilket säkerställer att eventuell senare framgång verkligen kommer från den nya träningen snarare än från dold skevhet i startdata.
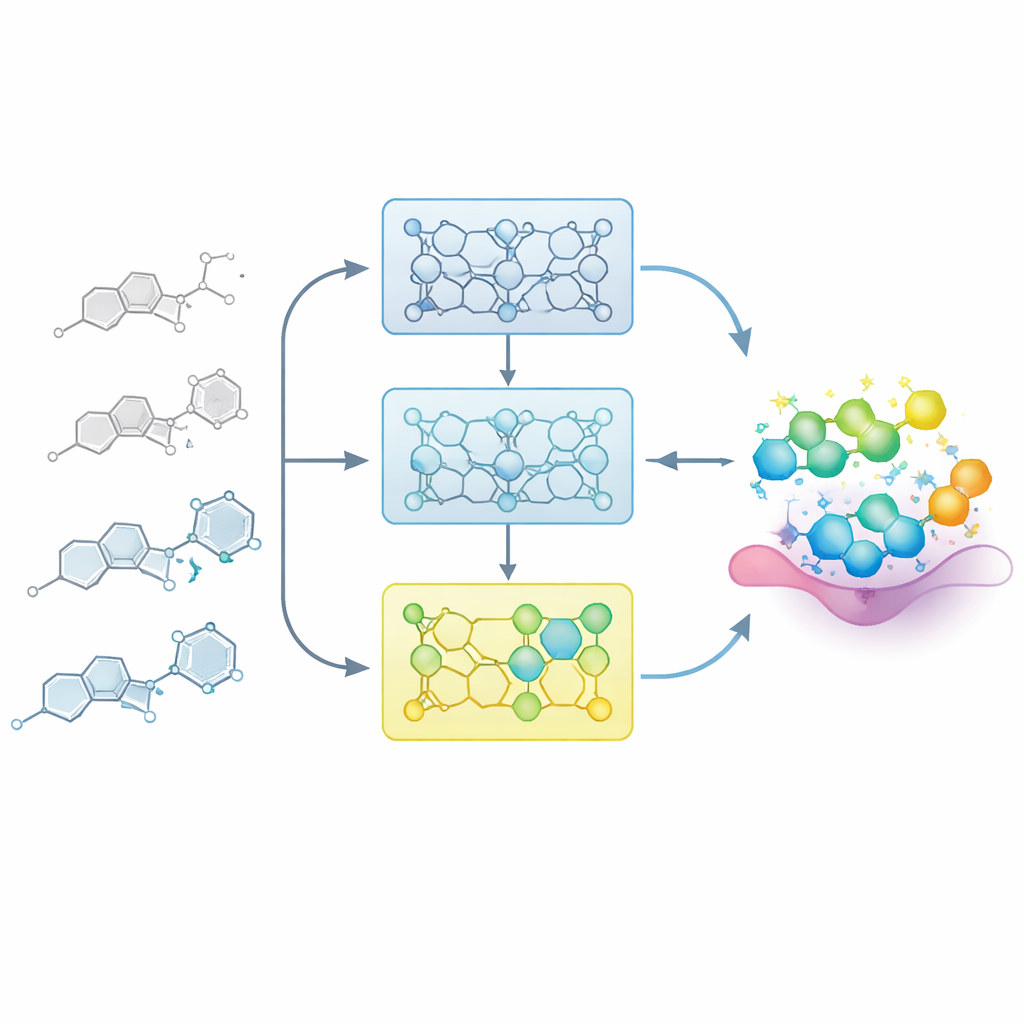
Låta modellen lära sig som en läkemedelskemist
I verkliga läkemedelsprojekt bygger kemister gradvis upp en karta mellan struktur och aktivitet: små förändringar i ett kärnscaffold kan göra en förening svagare eller starkare. Forskarlaget efterliknar denna process genom att mata modellen med noggrant ordnade serier av relaterade molekyler, kallade struktur–aktivitetssamband (SAR)-serier. Istället för att finjustera modellen i ett svep på alla kända exempel delar de upp varje serie i steg baserade på potens, från svagare till starkare medlemmar. Modellen exponeras först för de mindre aktiva föreningarna och finjusteras sedan successivt med delmängder som innehåller mer potenta exempel. Denna ”inkrementella träning” skapar en inlärningsbana där modellen varsamt vägleds mot den del av det kemiska rummet där de bästa molekylerna finns.
Från teori till nya, starkare läkemedelskandidater
För att testa om denna träningsstrategi verkligen hjälper kontrollerar teamet först om modellen kan ”återupptäcka” högaktiva molekyler som medvetet undanhållits från träningen. Med inkrementell träning genererar modellen topprankade design som matchar dessa dolda potenta föreningar mycket oftare än modeller som tränats i ett enda steg, vilket indikerar att den internaliserat de mönster som driver hög aktivitet. Författarna går sedan vidare till verklig design för två medicinskt relevanta mål: PPARγ, involverat i ämnesomsättning och inflammation, och RORγ, implicerat i immunreglering. Efter inkrementell träning på kända ligander för varje mål föreslår modellen nya analoger av utvalda scaffolds. När flera av dessa syntetiseras och testas i labbet visar sig alla nio PPARγ-design vara mycket potenta agonister, många långt överträffande den tidigare bästa molekylen, och en ny RORγ-design når nästan potensen hos den starkaste kända föreningen i sin serie samtidigt som den är strukturellt distinkt.
Vad detta innebär för framtida läkemedel
Genom att visa att en språkstilad modell inte bara kan uppfinna molekyler utan också förfina befintliga scaffolds för att överträffa de bästa kända exemplen — utan att luta sig mot externa poängsättningsverktyg — pekar detta arbete mot ett nytt sätt att bedriva läkemedelskemi. Den inkrementella träningsmetoden låter modellen absorbera subtila struktur–aktivitet-regler och deras långväga ömsesidiga beroenden, för att sedan förlänga dem in i outforskad terräng. För icke-specialister är huvudpoängen att AI nu kan agera mindre som en slumpmässig idégenerator och mer som en digitalt tränad kemists assistent, föreslå fokuserade, testbara förbättringar av lovande läkemedelsmolekyler och potentiellt snabba på vägen från tidiga träffar till optimerade läkemedel.
Citering: Hörmann, T., Mayer, D., Lewandowski, M. et al. Structural optimization of drug molecules with incrementally trained language models. Nat Commun 17, 3456 (2026). https://doi.org/10.1038/s41467-026-71591-w
Nyckelord: kemiska språkmodeller, de novo-läkemedelsdesign, struktur–aktivitetssamband, generativ kemi, AI inom läkemedelskemi