Clear Sky Science · it
Ottimizzazione strutturale delle molecole farmacologiche con modelli di linguaggio addestrati in modo incrementale
Insegnare ai computer a rimodellare i medicinali
I medicinali moderni spesso nascono come molecole promettenti ma imperfette che i chimici devono meticolosamente modificare per trasformarle in farmaci sicuri ed efficaci. Questo studio mostra come un sistema di intelligenza artificiale che «legge» le formule chimiche come una lingua possa imparare a svolgere autonomamente parte di questo lavoro di rifinitura, proponendo nuovi candidati farmacologici persino più potenti dei migliori esempi noti—senza fare affidamento su strumenti di scoring esterni o su tentativi ed errori basati su ipotesi.
Perché ottimizzare le molecole farmacologiche è così difficile
Una volta che i ricercatori individuano una molecola iniziale che agisce su un bersaglio biologico, inizia il vero lavoro: trasformare quel primo «hit» in qualcosa di potente, selettivo e adatto come medicinale. Tradizionalmente, i chimici progettano dozzine o centinaia di parenti stretti della struttura originale, li sintetizzano in laboratorio e testano ciascuno. Questi cicli di progettazione–sintesi–test richiedono anni di esperienza e grandi sforzi sperimentali. I metodi computazionali hanno cercato di aiutare, ma molti si concentrano su proprietà semplici, come quanto una molecola sia lipofila, invece che sull’effetto biologico complessivo che esercita. Altri approcci dipendono da strumenti di previsione separati («oracoli») che stimano l’attività e possono essere inaffidabili o non disponibili per molti bersagli.
Usare frasi chimiche per guidare la progettazione
Gli autori si basano sui modelli linguistici chimici, un tipo di sistema di deep learning che tratta le molecole come stringhe di caratteri (SMILES) e apprende la «grammatica» e i pattern che rendono una struttura chimicamente sensata e biologicamente interessante. Per prima cosa addestrano un modello su centinaia di migliaia di molecole bioattive note, escludendo deliberatamente tutto ciò che è collegato ai bersagli specifici che studieranno in seguito. Questo produce un modello generalista che comprende la chimica ma non ha conoscenza preventiva dei recettori scelti, garantendo che qualsiasi successo successivo derivi davvero dal nuovo addestramento e non da bias nascosti nei dati di partenza.
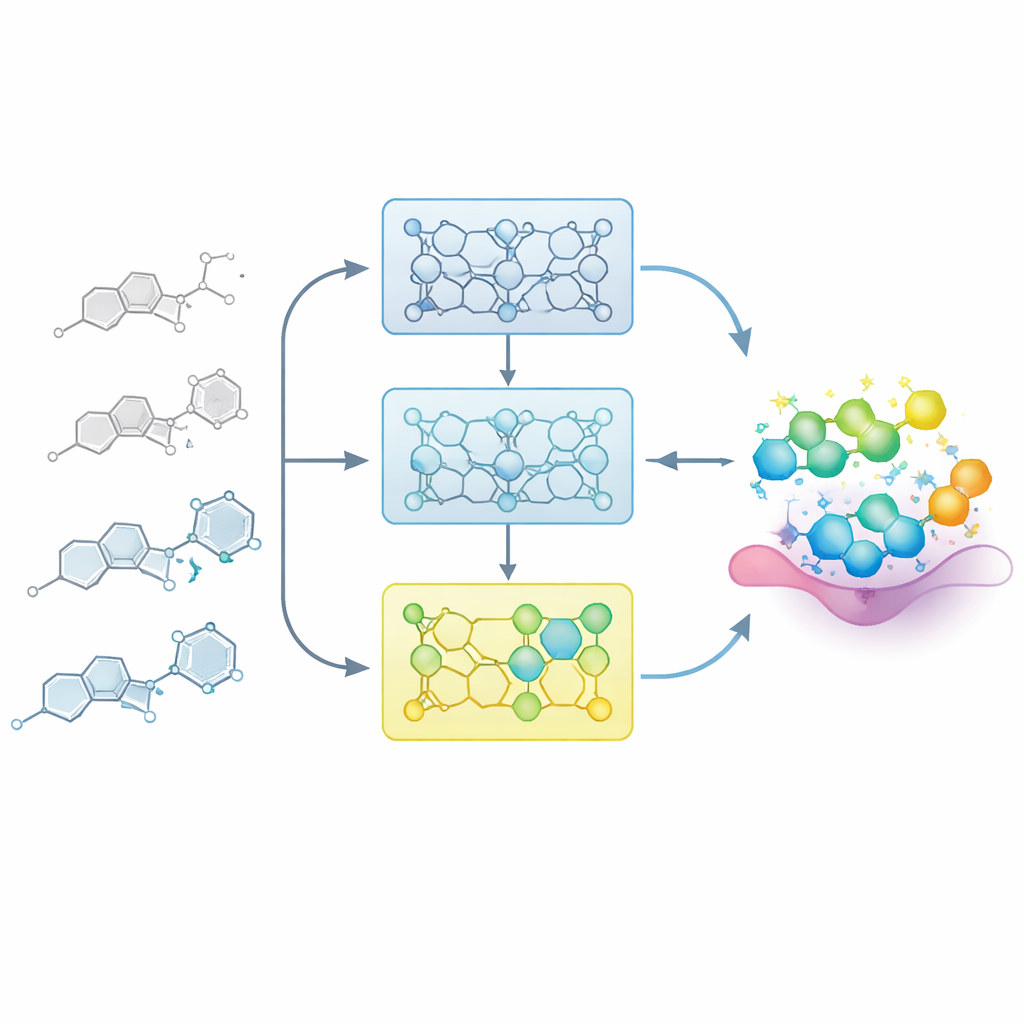
Lasciare che il modello apprenda come un chimico medicinale
Nei progetti farmacologici reali, i chimici costruiscono gradualmente una mappa tra struttura e attività: piccole modifiche a uno scaffold centrale possono rendere un composto più debole o più potente. I ricercatori imitano questo processo alimentando il modello con serie ordinate di molecole correlate, chiamate serie struttura–attività (SAR). Invece di adattare il modello in un’unica fase su tutti gli esempi conosciuti, suddividono ogni serie in passaggi basati sulla potenza, dai membri meno attivi a quelli più forti. Il modello viene prima esposto ai composti meno attivi, poi sottoposto a un fine-tuning successivo con sottoinsiemi contenenti esempi più potenti. Questo «addestramento incrementale» crea una traiettoria di apprendimento in cui il modello è gradualmente guidato verso la regione dello spazio chimico in cui risiedono le molecole migliori.
Dalla teoria a nuovi candidati farmacologici più potenti
Per verificare se questa strategia di addestramento sia davvero utile, il team controlla innanzitutto se il modello può «riscoprire» molecole altamente attive che sono state intenzionalmente escluse dall’addestramento. Con l’addestramento incrementale, il modello genera progetti di alta classifica che corrispondono a questi composti potenti nascosti molto più spesso rispetto ai modelli addestrati in un’unica fase, indicando che ha interiorizzato i pattern che guidano l’elevata attività. Gli autori passano poi alla progettazione nel mondo reale per due bersagli di rilevanza medica: PPARγ, coinvolto nel metabolismo e nell’infiammazione, e RORγ, implicato nella regolazione immunitaria. Dopo l’addestramento incrementale su ligandi noti per ciascun bersaglio, il modello propone nuovi analoghi di scaffold selezionati. Quando alcuni di questi vengono sintetizzati e testati in laboratorio, tutti e nove i progetti per PPARγ risultano essere agonisti altamente potenti, molti dei quali superano di gran lunga la migliore molecola precedente, e un nuovo progetto per RORγ si avvicina quasi alla potenza del composto più forte noto nella sua serie pur essendo strutturalmente distinto.
Cosa significa questo per i farmaci futuri
Dimostrando che un modello in stile linguistico può non solo inventare molecole ma anche perfezionare scaffold esistenti per superare i migliori esempi noti—senza appoggiarsi a strumenti di scoring esterni—questo lavoro indica una nuova modalità di fare chimica medicinale. L’approccio dell’addestramento incrementale permette al modello di assorbire regole sottili di struttura–attività e le loro interdipendenze a lungo raggio, quindi estenderle in territori inesplorati. Per i non specialisti, la conclusione principale è che l’IA può ora comportarsi meno come un generatore casuale di idee e più come l’assistente digitale di un chimico addestrato, proponendo miglioramenti mirati e verificabili a molecole promettenti e accelerando potenzialmente il percorso dagli hit iniziali ai farmaci ottimizzati.
Citazione: Hörmann, T., Mayer, D., Lewandowski, M. et al. Structural optimization of drug molecules with incrementally trained language models. Nat Commun 17, 3456 (2026). https://doi.org/10.1038/s41467-026-71591-w
Parole chiave: modelli linguistici chimici, progettazione di nuovi farmaci de novo, relazioni struttura–attività, chimica generativa, IA in chimica medicinale