Clear Sky Science · en
Structural optimization of drug molecules with incrementally trained language models
Teaching Computers to Tinker with Medicines
Modern medicines often begin as promising but imperfect molecules that chemists must painstakingly tweak to become safe and powerful drugs. This study shows how an artificial intelligence system that “reads” chemical formulas like a language can learn to carry out some of this tinkering on its own, proposing new drug candidates that are even more potent than the best known examples—without relying on external scoring tools or guesswork-heavy trial and error.
Why Optimizing Drug Molecules Is So Hard
Once researchers find an initial molecule that affects a biological target, the real work begins: turning that early “hit” into something strong, selective, and suitable as a medicine. Traditionally, chemists design dozens or hundreds of close relatives of the original structure, synthesize them in the lab, and test each one. These design–make–test cycles demand years of expertise and large experimental efforts. Computer methods have tried to help, but many focus on simple properties, such as how greasy a molecule is, rather than the full biological punch it delivers. Other methods depend on separate prediction tools (“oracles”) that estimate activity and can be unreliable or unavailable for many targets.
Using Chemical Sentences to Guide Design
The authors build on chemical language models, a type of deep learning system that treats molecules as strings of characters (SMILES) and learns the “grammar” and patterns that make a structure chemically sensible and biologically interesting. First, they pretrain a model on hundreds of thousands of known bioactive molecules, while deliberately filtering out anything related to the specific targets they later study. This produces a generalist model that understands chemistry but has no prior knowledge of the chosen receptors, ensuring that any later success truly comes from the new training it receives rather than from hidden bias in the starting data.
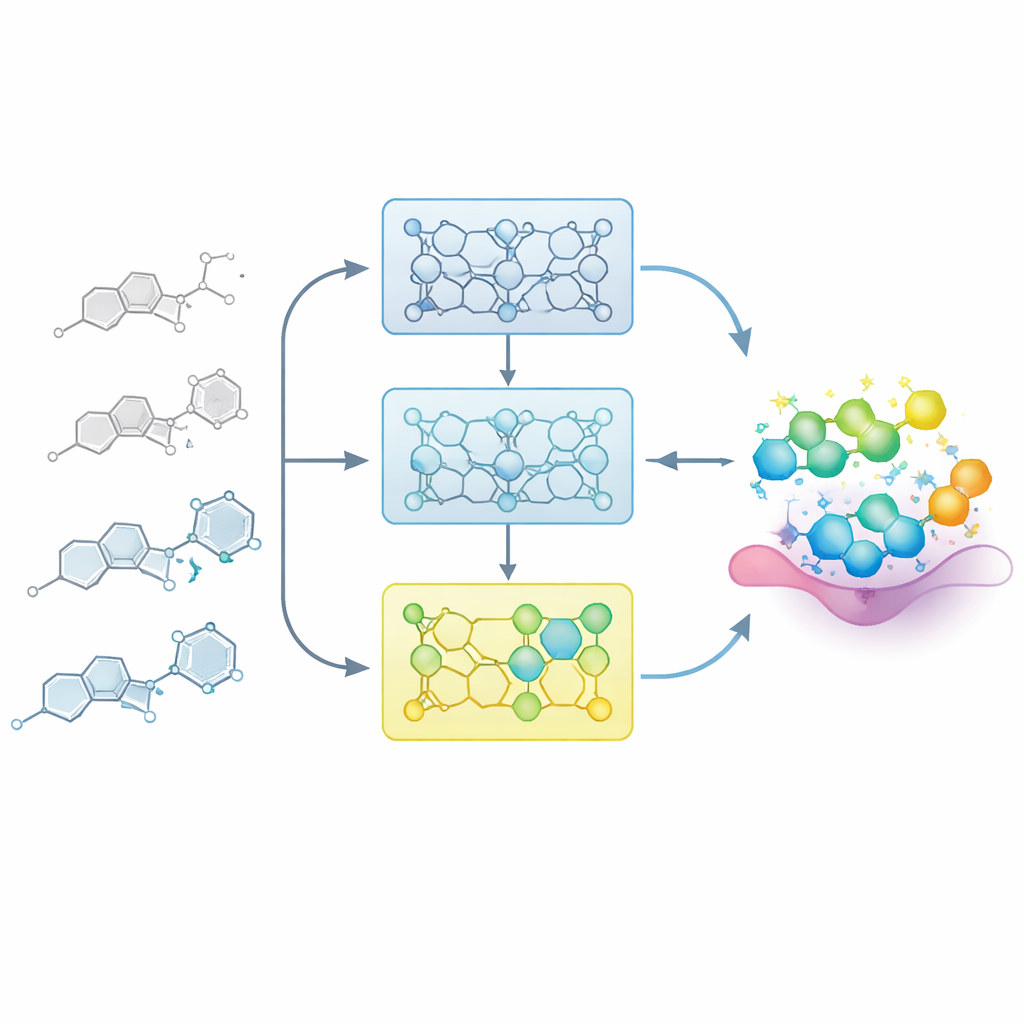
Letting the Model Learn Like a Medicinal Chemist
In real drug projects, chemists gradually build a map between structure and activity: small changes to a core scaffold can make a compound weaker or stronger. The researchers mimic this process by feeding the model carefully ordered series of related molecules, called structure–activity relationship (SAR) series. Instead of fine-tuning the model in one shot on all known examples, they split each series into steps based on potency, from weaker to stronger members. The model is first exposed to the less active compounds, then successively fine-tuned with subsets that contain more potent examples. This “incremental training” creates a learning trajectory in which the model is gently guided toward the region of chemical space where the best molecules reside.
From Theory to New, Stronger Drug Candidates
To test whether this training strategy truly helps, the team first checks whether the model can “rediscover” highly active molecules that were deliberately held back from training. With incremental training, the model generates top-ranked designs that match these hidden potent compounds far more often than models trained in a single step, indicating that it has internalized the patterns that drive high activity. The authors then move to real-world design for two medically relevant targets: PPARγ, involved in metabolism and inflammation, and RORγ, implicated in immune regulation. After incremental training on known ligands for each target, the model proposes new analogues of chosen scaffolds. When several of these are synthesized and tested in the lab, all nine PPARγ designs turn out to be highly potent agonists, many far surpassing the previous best molecule, and a new RORγ design nearly reaches the potency of the strongest known compound in its series while being structurally distinct.
What This Means for Future Medicines
By showing that a language-style model can not only invent molecules but also refine existing scaffolds to outperform the best known examples—without leaning on external scoring tools—this work points to a new way of doing medicinal chemistry. The incremental training approach lets the model absorb subtle structure–activity rules and their long-range interdependencies, then extend them into unexplored territory. For non-specialists, the key takeaway is that AI can now act less like a random idea generator and more like a digitally trained chemist’s assistant, proposing focused, testable improvements to promising drug molecules and potentially speeding the path from early hits to optimized medicines.
Citation: Hörmann, T., Mayer, D., Lewandowski, M. et al. Structural optimization of drug molecules with incrementally trained language models. Nat Commun 17, 3456 (2026). https://doi.org/10.1038/s41467-026-71591-w
Keywords: chemical language models, de novo drug design, structure–activity relationships, generative chemistry, AI in medicinal chemistry