Clear Sky Science · es
Optimización estructural de moléculas farmacéuticas con modelos de lenguaje entrenados de forma incremental
Enseñar a las computadoras a ajustar medicamentos
Los medicamentos modernos suelen empezar como moléculas prometedoras pero imperfectas que los químicos deben ajustar minuciosamente para convertirlas en fármacos seguros y eficaces. Este estudio muestra cómo un sistema de inteligencia artificial que “lee” fórmulas químicas como si fueran un lenguaje puede aprender a realizar parte de ese ajuste por sí mismo, proponiendo nuevos candidatos a fármaco que son incluso más potentes que los mejores ejemplos conocidos—sin depender de herramientas de puntuación externas ni de prueba y error basado en conjeturas.
Por qué es tan difícil optimizar moléculas farmacéuticas
Una vez que los investigadores encuentran una molécula inicial que afecta a un diana biológica, comienza el trabajo real: transformar ese primer “hit” en algo potente, selectivo y adecuado como fármaco. Tradicionalmente, los químicos diseñan docenas o cientos de parientes cercanos de la estructura original, los sintetizan en el laboratorio y prueban cada uno. Estos ciclos de diseño–fabricación–ensayo requieren años de experiencia y grandes esfuerzos experimentales. Los métodos computacionales han intentado ayudar, pero muchos se centran en propiedades simples, como la lipofilia de una molécula, en lugar del efecto biológico completo que produce. Otros métodos dependen de herramientas predictivas separadas (“oráculos”) que estiman la actividad y que pueden ser poco fiables o no estar disponibles para muchos dianas.
Usar frases químicas para guiar el diseño
Los autores se apoyan en modelos de lenguaje químico, un tipo de sistema de aprendizaje profundo que trata las moléculas como cadenas de caracteres (SMILES) y aprende la “gramática” y los patrones que hacen que una estructura sea químicamente coherente e interesantemente biológica. Primero, preentrenan un modelo con cientos de miles de moléculas bioactivas conocidas, filtrando deliberadamente todo lo relacionado con las dianas específicas que estudiarán después. Esto produce un modelo generalista que entiende la química pero que no tiene conocimiento previo de los receptores escogidos, garantizando que cualquier éxito posterior proceda realmente del nuevo entrenamiento y no de un sesgo oculto en los datos iniciales.
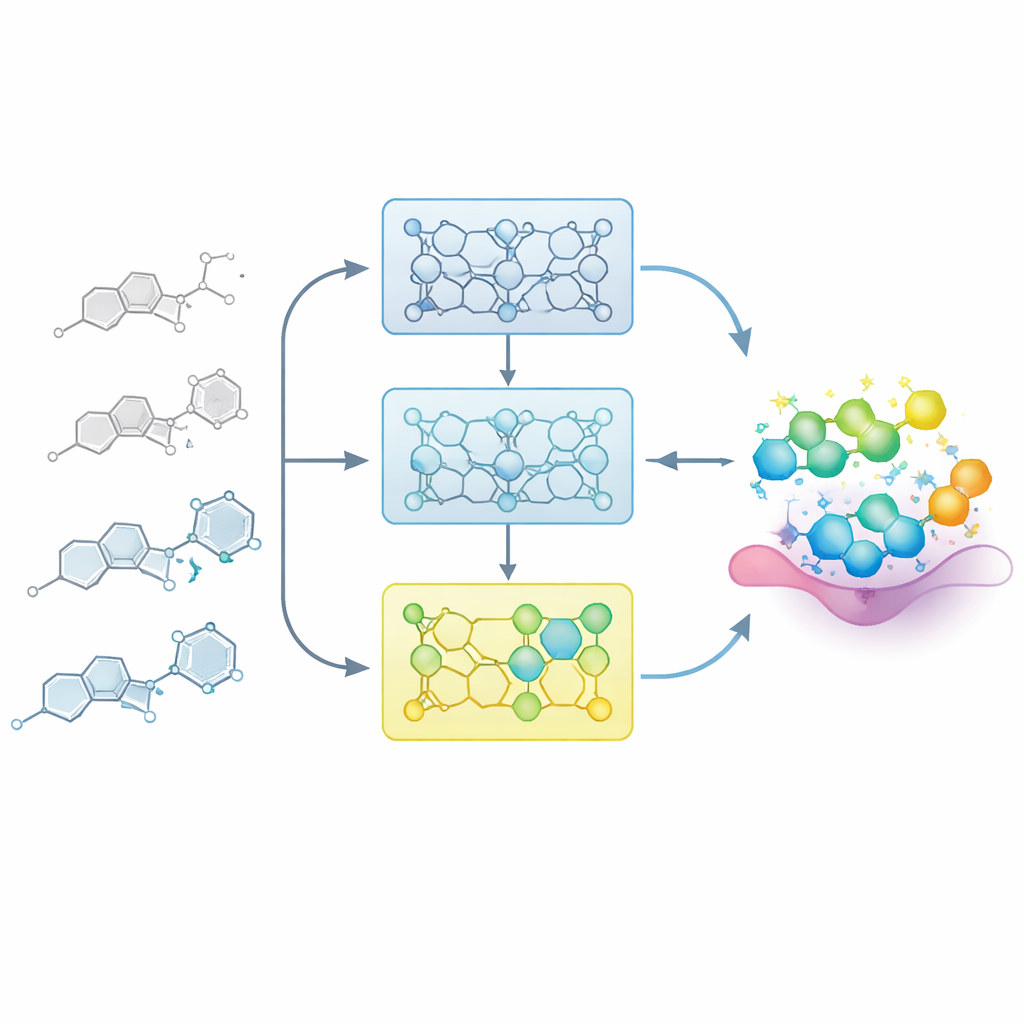
Permitir que el modelo aprenda como un químico medicinal
En proyectos farmacéuticos reales, los químicos construyen gradualmente un mapa entre estructura y actividad: pequeños cambios en un andamiaje central pueden debilitar o reforzar un compuesto. Los investigadores imitan este proceso alimentando al modelo con series ordenadas de moléculas relacionadas, llamadas series de relación estructura–actividad (SAR). En lugar de ajustar el modelo de una sola vez con todos los ejemplos conocidos, dividen cada serie en pasos basados en la potencia, desde los miembros más débiles hasta los más potentes. El modelo se expone primero a los compuestos menos activos y luego se ajusta sucesivamente con subconjuntos que contienen ejemplos más potentes. Este “entrenamiento incremental” crea una trayectoria de aprendizaje en la que el modelo es guiado suavemente hacia la región del espacio químico donde residen las mejores moléculas.
De la teoría a nuevos candidatos a fármaco más potentes
Para comprobar si esta estrategia de entrenamiento realmente ayuda, el equipo primero verifica si el modelo puede “redescubrir” moléculas altamente activas que se reservaron deliberadamente fuera del entrenamiento. Con el entrenamiento incremental, el modelo genera diseños mejor clasificados que coinciden con estos compuestos ocultos y potentes con mucha más frecuencia que los modelos entrenados en un solo paso, lo que indica que ha internalizado los patrones que impulsan la alta actividad. Los autores pasan luego al diseño del mundo real para dos dianas de relevancia médica: PPARγ, implicada en el metabolismo y la inflamación, y RORγ, implicada en la regulación inmunitaria. Tras el entrenamiento incremental con ligandos conocidos para cada diana, el modelo propone nuevos análogos de andamiajes seleccionados. Cuando varios de estos se sintetizan y prueban en el laboratorio, los nueve diseños para PPARγ resultan ser agonistas altamente potentes, muchos superando con creces la mejor molécula previa, y un nuevo diseño para RORγ alcanza casi la potencia del compuesto más fuerte conocido en su serie siendo, además, estructuralmente distinto.
Qué significa esto para los futuros medicamentos
Al demostrar que un modelo de estilo lingüístico no solo puede inventar moléculas sino también refinar andamiajes existentes para superar los mejores ejemplos conocidos—sin apoyarse en herramientas de puntuación externas—este trabajo apunta a una nueva forma de hacer química medicinal. El enfoque de entrenamiento incremental permite al modelo absorber reglas sutiles de relación estructura–actividad y sus interdependencias a largo alcance, y luego extenderlas hacia territorio inexplorado. Para no especialistas, la conclusión clave es que la IA puede actuar ahora menos como un generador aleatorio de ideas y más como el asistente digitalmente entrenado de un químico, proponiendo mejoras enfocadas y comprobables a moléculas prometedoras y potencialmente acelerando el camino desde los primeros hits hasta fármacos optimizados.
Cita: Hörmann, T., Mayer, D., Lewandowski, M. et al. Structural optimization of drug molecules with incrementally trained language models. Nat Commun 17, 3456 (2026). https://doi.org/10.1038/s41467-026-71591-w
Palabras clave: modelos de lenguaje químicos, diseño de fármacos de novo, relaciones estructura–actividad, química generativa, IA en química medicinal