Clear Sky Science · ja
漸進的に訓練された言語モデルによる医薬分子の構造最適化
薬を“いじる”ことをコンピュータに教える
現代の医薬品は、多くの場合、有望だが不完全な分子から始まり、化学者が安全で効果的な薬に仕上げるために手作業で慎重に調整します。本研究は、化学式を言語のように「読み取る」人工知能システムがそのような調整を自律的に学び、既知の最良例を上回る強力な新候補分子を外部の評価ツールや推測に頼ることなく提案できることを示しています。
なぜ医薬分子の最適化は難しいのか
研究者が生物学的標的に影響する初期分子を見つけると、本当の仕事が始まります:その初期の「ヒット」を強力で選択的、かつ医薬品として適したものに変えることです。従来は化学者が元の構造の多数の近縁体を設計し、合成し、それぞれを試験してきました。こうした設計–作製–試験のサイクルは何年にも及ぶ専門知識と大規模な実験努力を要します。計算手法も支援を試みてきましたが、多くは分子の疎水性など単純な性質に注目しがちで、完全な生物学的効果を捉えられていません。別の手法は活性を推定する外部の予測ツール(「オラクル」)に依存しますが、これらは信頼性が低かったり、多くの標的では利用できなかったりします。
化学の文を設計に活かす
著者らは、分子を文字列(SMILES)として扱い、化学的に妥当で生物学的に興味深い構造を生む「文法」やパターンを学習する一種の深層学習システムである化学言語モデルに基づいています。まず、彼らは数十万件の既知の生物活性分子でモデルを事前学習させますが、後に調べる特定の標的に関連するものは意図的に除外します。これにより、化学を理解する一般モデルが得られ、選んだ受容体に関する先入観がない状態になるため、その後の成功が出発データの隠れたバイアスではなく、新たな学習によるものであることが保証されます。
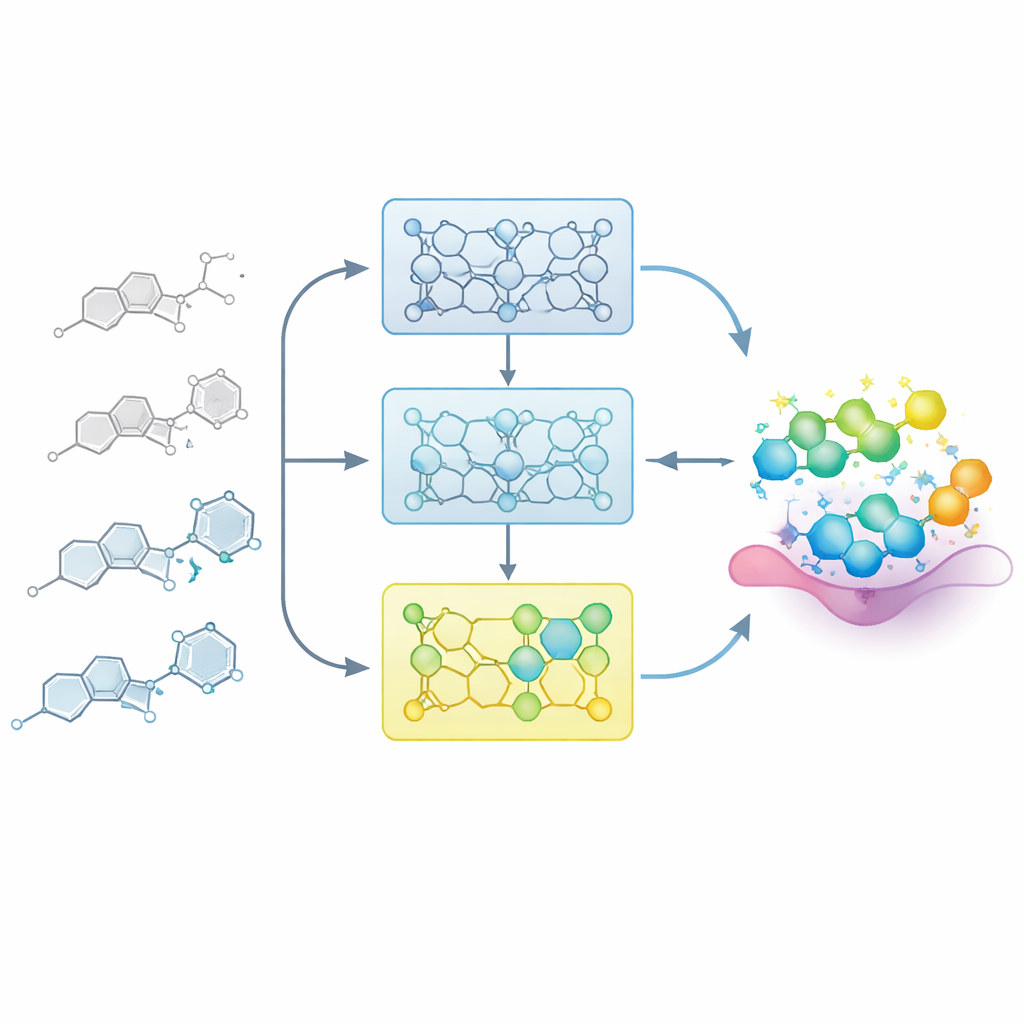
創薬化学者のようにモデルを学習させる
実際の医薬プロジェクトでは、化学者は構造と活性の間の地図を段階的に作り上げます:コア骨格に対する小さな変更が化合物を弱めたり強めたりします。研究者らは、構造–活性相関(SAR)シリーズと呼ばれる関連分子の慎重に並べられた系列をモデルに与えることでこのプロセスを模倣します。既知の例すべてで一度にファインチューニングする代わりに、彼らは各シリーズを活性に基づく段階に分割し、弱いものから強いものへと並べます。モデルはまず活性の低い化合物に触れ、次により活性の高い例を含む部分集合で順次ファインチューニングされます。この「漸進的訓練」は、モデルを最良の分子が存在する化学空間の領域へと穏やかに導く学習軌道を作ります。
理論から新しい、より強力な候補へ
この訓練戦略が本当に有効かを検証するため、研究チームはまずモデルが訓練から意図的に除外した高活性分子を「再発見」できるかどうかを確認します。漸進的訓練を行うと、モデルはこれらの隠された高活性化合物に一致する上位設計を一段と多く生成し、単一ステップで訓練したモデルよりも高活性を生み出すパターンを内在化していることを示します。著者らは次に、実際の医療的に関連する二つの標的、代謝や炎症に関与するPPARγと免疫制御に関係するRORγに対する設計に移ります。各標的の既知リガンドで漸進的に訓練した後、モデルは選んだ骨格の新たな類似体を提案します。いくつかが合成・試験されたところ、PPARγに対する9件すべての設計が非常に高いアゴニスト活性を示し、多くが従来の最良分子を大きく上回りました。また、RORγの新しい設計は系列内の最も強力な既知化合物の活性にほぼ達しつつ、構造的には異なるものでした。
将来の医薬品にとっての意味
言語スタイルのモデルが分子を発明するだけでなく、既存の骨格を改良して既知の最良例を上回ることができ、しかも外部の評価ツールに依存しないことを示した本研究は、医薬化学の新しいあり方を示唆します。漸進的訓練アプローチは、モデルに微妙な構造–活性規則やそれらの長距離の相互依存性を吸収させ、それを未踏の領域へ拡張させます。専門外の人への要点は、AIがもはや単なるランダムなアイデア生成器ではなく、デジタルに訓練された化学者の助手のように振る舞い、有望な分子に対して焦点を絞った試験可能な改良案を提案し、初期ヒットから最適化医薬品への道を速める可能性があるということです。
引用: Hörmann, T., Mayer, D., Lewandowski, M. et al. Structural optimization of drug molecules with incrementally trained language models. Nat Commun 17, 3456 (2026). https://doi.org/10.1038/s41467-026-71591-w
キーワード: 化学言語モデル, デ・ノボ医薬品設計, 構造–活性相関, 生成的化学, 医薬化学におけるAI