Clear Sky Science · nl
Structurele optimalisatie van geneesmiddelmoleculen met incrementeel getrainde taalmodellen
Computers leren sleutelen aan medicijnen
Moderne geneesmiddelen beginnen vaak als veelbelovende maar onvolmaakte moleculen die chemici zorgvuldig moeten bijstellen om veilige en krachtige medicijnen te worden. Deze studie toont hoe een kunstmatig-intelligentiesysteem dat chemische formules als een taal "leest" kan leren om een deel van dat bijstelwerk zelfstandig uit te voeren, en nieuwe medicijnkandidaten kan voorstellen die zelfs krachtiger zijn dan de beste bekende voorbeelden—zonder te steunen op externe scoringshulpmiddelen of op giswerk en veelvuldige trial-and-error.
Waarom het optimaliseren van geneesmiddelmoleculen zo moeilijk is
Zodra onderzoekers een initiëel molecuul vinden dat een biologisch doelwit beïnvloedt, begint het echte werk: van die eerste "hit" iets maken dat sterk, selectief en geschikt is als geneesmiddel. Traditioneel ontwerpen chemici tientallen tot honderden nauwe varianten van de oorspronkelijke structuur, synthetiseren die in het lab en testen elk exemplaar. Deze ontwerp–maak–test-cycli vergen jaren aan expertise en grote experimentele inspanningen. Computermethoden hebben geprobeerd te helpen, maar veel richten zich op eenvoudige eigenschappen, zoals hoe vetachtig een molecuul is, in plaats van op de volledige biologische effectiviteit. Andere methoden hangen af van afzonderlijke voorspellingstools ("orakels") die activiteit schatten maar onbetrouwbaar kunnen zijn of voor veel doelwitten niet beschikbaar zijn.
Chemische zinnen gebruiken om ontwerp te sturen
De auteurs bouwen voort op chemische taalmodellen, een type deep learning-systeem dat moleculen behandelt als tekenreeksen (SMILES) en leert welke "grammatica" en patronen een structuur chemisch zinvol en biologisch relevant maken. Eerst pretrainen ze een model op honderden duizenden bekende bioactieve moleculen, terwijl ze doelbewust alles filteren dat gerelateerd is aan de specifieke doelwitten die ze later bestuderen. Dit levert een generalistisch model op dat chemie begrijpt maar geen voorkennis heeft van de gekozen receptoren, waardoor elk later succes daadwerkelijk voortkomt uit de nieuwe training en niet uit verborgen vooringenomenheid in de startdata.
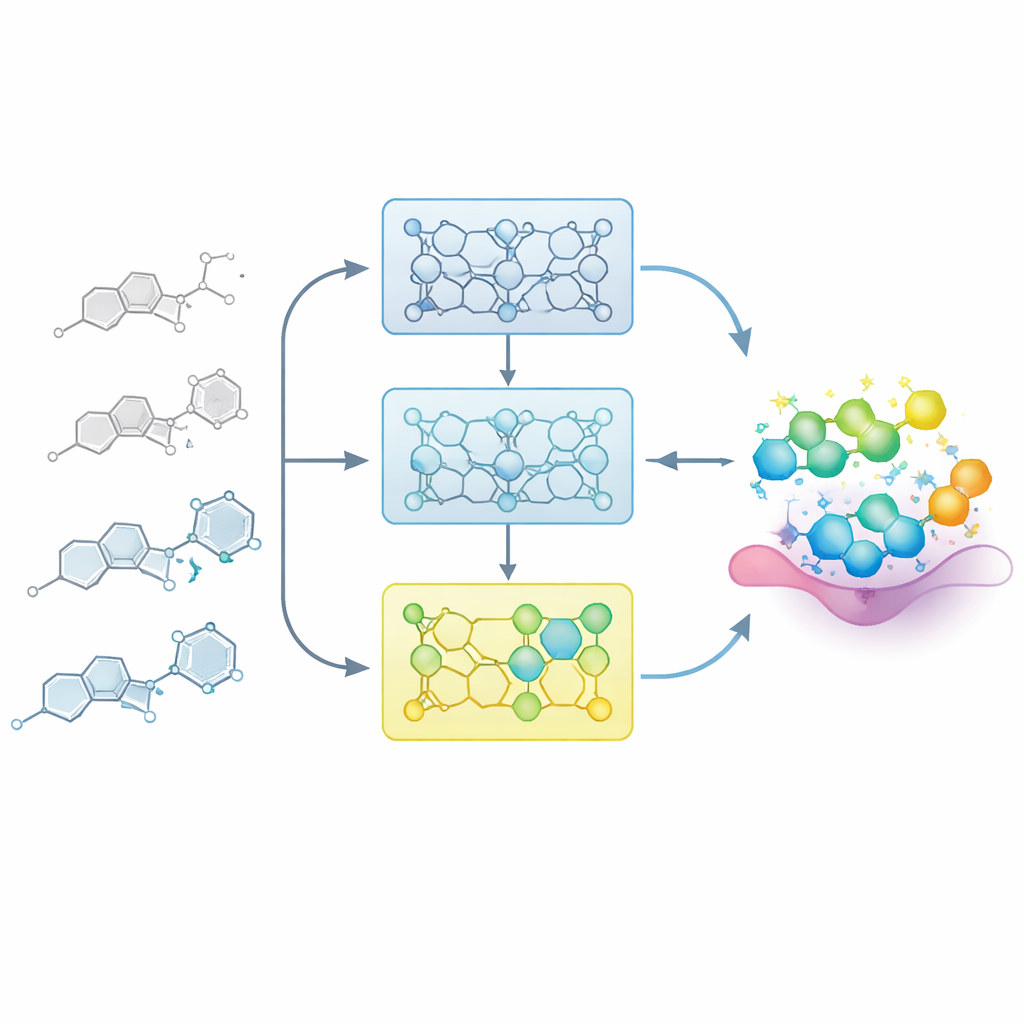
Het model leren als een medicinale chemicus
In echte medicijnprojecten bouwen chemici geleidelijk een kaart tussen structuur en activiteit: kleine veranderingen aan een basisskelet kunnen een verbinding zwakker of sterker maken. De onderzoekers bootsen dit proces na door het model zorgvuldig geordende reeksen van verwante moleculen te voeden, zogenaamde structuur–activiteit-relaties (SAR)-reeksen. In plaats van het model in één keer bij te stellen op alle bekende voorbeelden, splitsen ze elke reeks op in stappen op basis van potentie, van zwakkere naar sterkere leden. Het model wordt eerst blootgesteld aan de minder actieve verbindingen en vervolgens stapsgewijs bijgetraind met subsets die meer potente voorbeelden bevatten. Deze "incrementele training" creëert een leertraject waarin het model zachtjes wordt geleid naar het deel van de chemische ruimte waar de beste moleculen zich bevinden.
Van theorie naar nieuwe, sterkere medicijnkandidaten
Om te testen of deze trainingsstrategie daadwerkelijk helpt, controleert het team eerst of het model hoogactieve moleculen kan "herontdekken" die bewust uit de training werden weggelaten. Met incrementele training genereert het model topgerangschikte ontwerpen die veel vaker overeenkomen met deze verborgen potente verbindingen dan modellen die in één stap zijn getraind, wat aangeeft dat het de patronen die hoge activiteit aandrijven heeft geïnternaliseerd. De auteurs verschuiven daarna naar ontwerpen voor twee medisch relevante doelwitten: PPARγ, betrokken bij metabolisme en ontsteking, en RORγ, geïmpliceerd in immuunregulatie. Na incrementele training op bekende liganden voor elk doelwit stelt het model nieuwe analogen van gekozen skeletten voor. Wanneer verschillende hiervan worden gesynthetiseerd en in het laboratorium getest, blijken alle negen PPARγ-ontwerpen zeer potente agonisten te zijn, waarvan vele de eerdere beste molecule ver overtreffen, en één nieuw RORγ-ontwerp benadert bijna de potentie van de sterkste bekende verbinding in die reeks terwijl het structureel onderscheidend is.
Wat dit betekent voor toekomstige geneesmiddelen
Door te laten zien dat een taalachtig model niet alleen moleculen kan verzinnen maar ook bestaande skeletten kan verfijnen tot beter presterende voorbeelden—zonder te leunen op externe scoringshulpmiddelen—wijst dit werk op een nieuwe manier van medicinale chemie bedrijven. De incrementele trainingsaanpak laat het model subtiele structuur–activiteitsregels en hun verre afhankelijkheden absorberen en deze vervolgens uitbreiden naar onontgonnen gebieden. Voor niet-specialisten is de kernboodschap dat AI nu minder op een willekeurige ideeëngenerator lijkt en meer op de digitaal getrainde assistent van een chemicus, die gerichte, testbare verbeteringen aan veelbelovende medicijnmoleculen voorstelt en mogelijk het pad van vroege hits naar geoptimaliseerde geneesmiddelen versnelt.
Bronvermelding: Hörmann, T., Mayer, D., Lewandowski, M. et al. Structural optimization of drug molecules with incrementally trained language models. Nat Commun 17, 3456 (2026). https://doi.org/10.1038/s41467-026-71591-w
Trefwoorden: chemische taalmodellen, de novo medicijnontwerp, structuur–activiteitsrelaties, generatieve chemie, AI in medicinale chemie