Clear Sky Science · tr
Artırımlı olarak eğitilmiş dil modelleriyle ilaç moleküllerinin yapısal optimizasyonu
Bilgisayarlara İlaçlarla Oynamayı Öğretmek
Modern ilaçlar genellikle ümit vaat eden ama kusurlu moleküller olarak başlar ve kimyagerlerin bunları güvenli ve etkili ilaçlara dönüştürmek için titizlikle ince ayar yapması gerekir. Bu çalışma, kimyasal formülleri bir dil gibi “okuyan” yapay zeka sisteminin bu tür ince ayarları kendi başına yapmayı öğrenebileceğini; harici puanlama araçlarına veya yoğun deneme–yanılma tahminlerine dayanmayarak, bilinen en iyi örneklerden daha etkili yeni ilaç adayları önerebildiğini gösteriyor.
İlaç Moleküllerini Optimize Etmenin Zorluğu
Araştırmacılar bir biyolojik hedefi etkileyen ilk molekülü bulduğunda, asıl iş başlar: o ilk “hit”i güçlü, seçici ve ilaç olarak uygun hale getirmek. Geleneksel olarak kimyagerler, orijinal yapıya yakın onlarca veya yüzlerce akraba tasarlar, bunları laboratuvarda sentezler ve her birini test eder. Bu tasarım–yapım–test döngüleri yıllarca süren uzmanlık ve büyük deneysel çabalar gerektirir. Bilgisayar yöntemleri yardımcı olmaya çalıştı, ancak birçoğu molekülün yağlılık gibi basit özelliklerine odaklanır, oysa tam biyolojik etkinliğin tamamını yakalamak zordur. Diğer yaklaşımlar, etkinliği tahmin eden ayrı “kâhin” araçlarına dayanır; bunlar birçok hedef için güvenilmez olabilir veya bulunmayabilir.
Tasarımı Yönlendirmek İçin Kimyasal Cümleleri Kullanmak
Yazarlar, molekülleri karakter dizileri (SMILES) olarak ele alan ve bir yapıyı kimyasal açıdan mantıklı ve biyolojik açıdan ilginç kılan “dilbilgisini” ve kalıpları öğrenen bir tür derin öğrenme sistemi olan kimyasal dil modellerinin üzerine inşa ediyor. Öncelikle, daha sonra inceledikleri özel hedeflerle ilgili herhangi bir şeyi kasten filtreleyerek yüz binlerce bilinen biyolojik açıdan aktif molekül üzerinde bir modeli önceden eğitiyorlar. Bu, kimyayı anlayan ama seçilen reseptörler hakkında önceden bilgi taşımayan genel amaçlı bir model üretir; böylece sonraki başarıların gerçekten yeni eğitimden kaynaklandığı, başlangıç verilerindeki gizli önyargılardan değil.
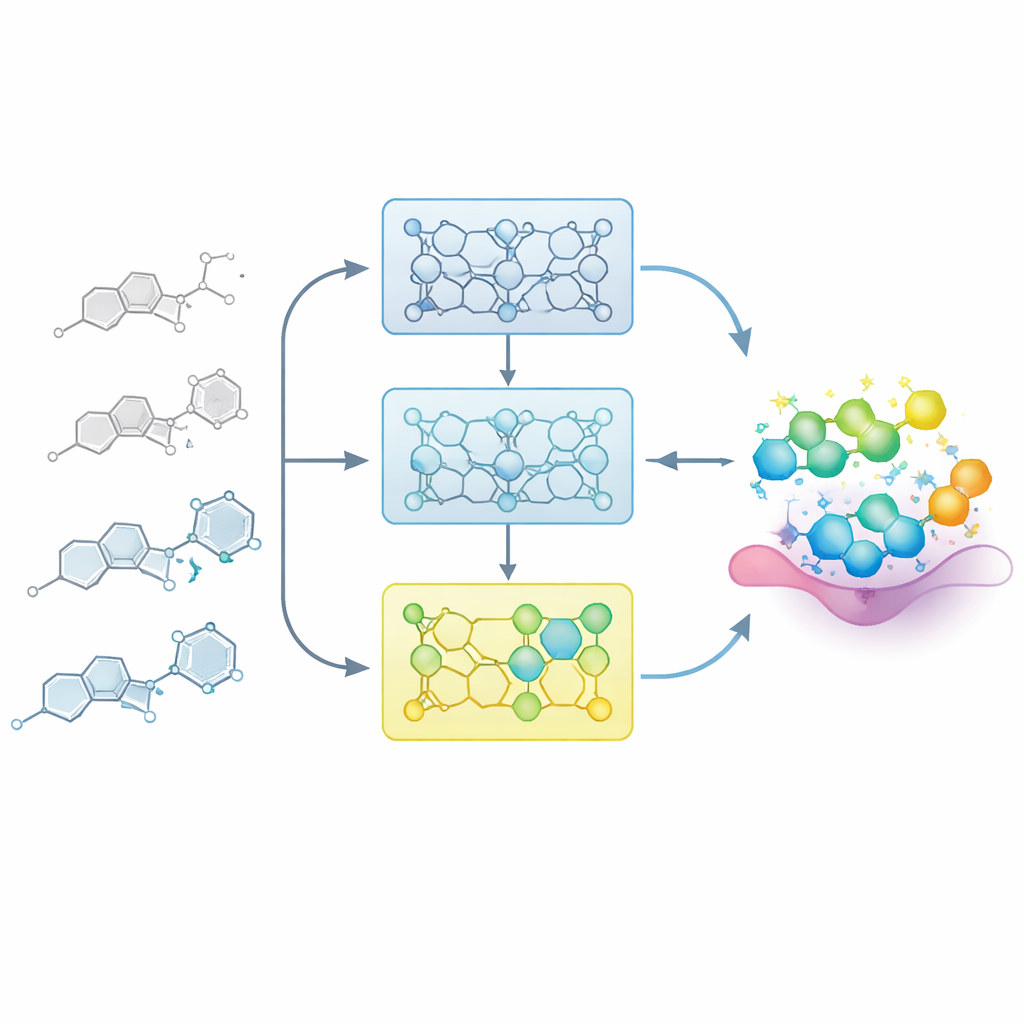
Modelin Bir İlaç Kimyageri Gibi Öğrenmesine İzin Vermek
Gerçek ilaç projelerinde kimyagerler yapıyla etkinlik arasında kademeli olarak bir harita oluşturur: bir çekirdek iskeletteki küçük değişiklikler bir bileşiği zayıflatabilir veya güçlendirebilir. Araştırmacılar bu süreci, modele yapı–etkinlik ilişkisi (SAR) dizileri adı verilen dikkatle sıralanmış ilgili molekül serilerini vererek taklit ediyor. Modeli bilinen tüm örnekler üzerinde tek seferde ince ayar yapmak yerine, her seriyi etkinliğe göre daha zayıftan daha güçlüye adım adım bölüyorlar. Model önce daha az aktif bileşiklere maruz bırakılıyor, ardından giderek daha güçlü örnekler içeren alt kümelerle ardışık ince ayarlar yapılıyor. Bu “artırımlı eğitim” modeli, en iyi moleküllerin bulunduğu kimyasal alan bölgesine nazikçe yönlendiren bir öğrenme yolu oluşturuyor.
Teoriden Yeni, Daha Güçlü İlaç Adaylarına
Bu eğitim stratejisinin gerçekten yardımcı olup olmadığını test etmek için ekip önce modelin eğitimden kasıtlı olarak geri tutulmuş yüksek etkinlikli molekülleri “yeniden keşfedip” keşfedemeyeceğini kontrol ediyor. Artırımlı eğitimle model, tek adımda eğitilmiş modellere kıyasla bu gizli güçlü bileşiklerle çok daha sık eşleşen üst sıradaki tasarımlar üretiyor; bu, yüksek etkinliği yönlendiren kalıpları içselleştirdiğini gösteriyor. Yazarlar ardından metabolizma ve inflamasyonda rol oynayan PPARγ ile bağışıklık düzenlemesinde rolü olan RORγ adlı iki tıbbi açıdan ilgili hedef için gerçek dünya tasarımına geçiyor. Her hedef için bilinen ligandlar üzerinde artırımlı eğitim yapıldıktan sonra model, seçili iskeletlerin yeni analoqlarını öneriyor. Bunlardan birkaç tanesi sentezlenip laboratuvarda test edildiğinde, dokuz PPARγ tasarımının tamamı yüksek güçlü agonistler olarak ortaya çıkıyor; birçoğu önceki en iyi molekülü büyük ölçüde geride bırakıyor ve yeni bir RORγ tasarımı da serisindeki en güçlü bilinen bileşiğin potansiyeline neredeyse ulaşıyor, üstelik yapısal olarak farklı bir düzen gösteriyor.
Geleceğin İlaçları İçin Anlamı
Bir dil-stili modelinin yalnızca moleküller icat etmekle kalmayıp mevcut iskeletleri bilinen en iyi örneklerin ötesine taşıyacak şekilde rafine edebileceğini—harici puanlama araçlarına dayanmayarak—göstermesi, ilaç kimyası yapımında yeni bir yol öneriyor. Artırımlı eğitim yaklaşımı modelin ince yapı–etkinlik kurallarını ve bunların uzun menzilli karşılıklı bağımlılıklarını emmesine izin veriyor, ardından bunları keşfedilmemiş bölgelere genişletebiliyor. Uzman olmayanlar için temel çıkarım şu: Yapay zeka artık rastgele fikir üreten bir sistemden çok, dijital olarak eğitilmiş bir kimyager yardımcısı gibi davranabiliyor; ümit veren ilaç moleküllerine odaklanmış, test edilebilir iyileştirmeler önererek erken hitlerden optimize edilmiş ilaçlara giden yolu hızlandırma potansiyeline sahip.
Atıf: Hörmann, T., Mayer, D., Lewandowski, M. et al. Structural optimization of drug molecules with incrementally trained language models. Nat Commun 17, 3456 (2026). https://doi.org/10.1038/s41467-026-71591-w
Anahtar kelimeler: kimyasal dil modelleri, de novo ilaç tasarımı, yapı–etkinlik ilişkileri, üretken kimya, ilaç kimyasında yapay zeka