Clear Sky Science · zh
LMO7介导的POLR2A降解通过MDM4/p53/p21轴促进细胞衰老
为什么衰老细胞重要
随着年龄增长,我们的细胞逐渐失去分裂与修复组织的能力。许多功能受损的细胞并不会死亡;相反,它们进入一种长期的“睡眠”状态,称为细胞衰老。衰老细胞通过释放炎性因子削弱器官功能并驱动与年龄相关的疾病。本研究提出了一个看似简单却重要的问题:是什么使健康细胞转向这种衰老状态——以及能否控制这一开关?
细胞复制室里的关键工人
在每个细胞内,有一个大型分子机器RNA聚合酶II读取DNA并帮助启动基因表达。POLR2A是该机器中最大且最重要的部分。早期研究提示POLR2A在老化组织和早衰疾病中下降,但尚不清楚这是老化的副产物还是成因之一。作者研究了人类成纤维细胞(一种常见的皮肤和肺部支持细胞),并观察了不同年龄小鼠的组织,以了解POLR2A随时间的变化。
在实验室中观察细胞如何变老
团队用了两种方法在培养皿中诱导细胞衰老。在“复制性”衰老模型中,让成纤维细胞不断分裂,直到它们自然减缓。在“应激诱导”衰老模型中,年轻细胞短暂暴露于过氧化氢,导致氧化损伤。在两种模型中,越来越多的细胞表现出典型的衰老标志:停止分裂、在标准衰老标记物上呈阳性并释放炎性分子。显著的是,随着这些变化出现,POLR2A蛋白水平急剧下降,而其mRNA并未相应减少,表明是蛋白被破坏而非转录减少。类似的POLR2A下降也在老年小鼠的大脑、心脏和肺中观察到,将这一过程与体内老化联系起来。
失去POLR2A如何触发衰老开关
为检验因果关系,研究人员在年轻成纤维细胞中刻意降低POLR2A水平。细胞迅速呈现出衰老表型:停止分裂、衰老标记物阳性并分泌炎性信号。在分子层面上,一个围绕p53及其伙伴p21构建的知名保护通路被强烈激活。当同时敲低POLR2A和p53时,细胞大多逃脱了衰老,表明POLR2A通常通过这一p53–p21途径帮助抑制衰老。进一步挖掘发现,p53水平升高并非因为合成增多,而是因为降解变慢。一个名为MDM4的蛋白——通常帮助标记p53以便降解——在POLR2A下降时也减少,导致p53更稳定并更强烈地传递阻止细胞分裂的信号。
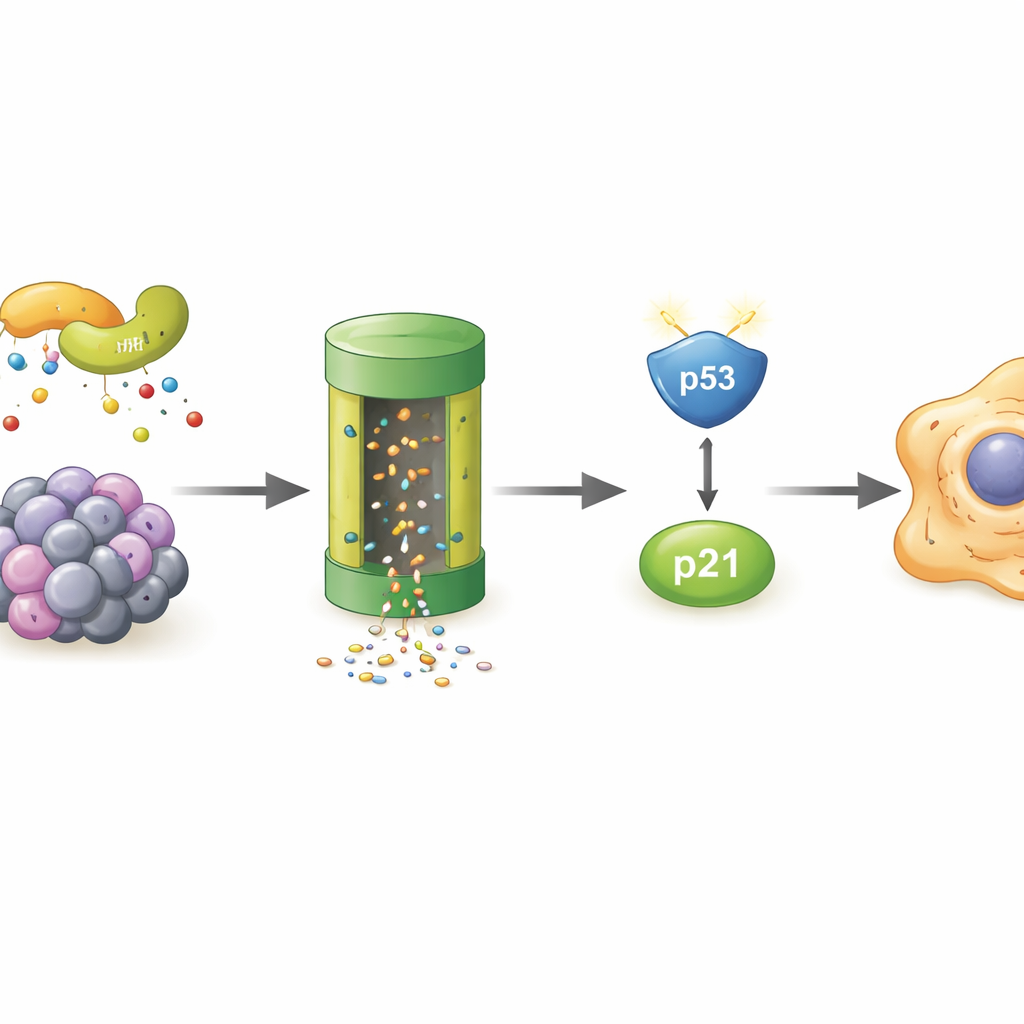
是谁在衰老细胞中破坏POLR2A
既然POLR2A在衰老细胞中被更快地清除,作者便寻找负责的细胞“垃圾系统”。他们证明阻断蛋白酶体——细胞主要的蛋白降解机器——可以挽救POLR2A水平,并且POLR2A带有更多的泛素标记,泛素是靶向蛋白降解的分子旗帜。一次质谱筛选指出LMO7,这是一个支架蛋白,可作为添加这些标记的酶复合体的一部分。在受压细胞中,LMO7与POLR2A的结合更强。当去除LMO7时,POLR2A不再被大量泛素化,其水平即使在氧化应激下也保持更高,明确将LMO7指认为细胞衰老过程中导致POLR2A丧失的核心驱动因子。
在细胞内部把时钟拨回去
最后,研究者探问提升POLR2A是否能抵抗衰老。他们使用CRISPR激活技术,温和地上调细胞自身的POLR2A基因,而不是外源性加入拷贝。POLR2A升高的细胞对应激诱导和复制性衰老更具抵抗力:较少细胞呈衰老标记物阳性,更多细胞继续分裂。与此同时,MDM4水平保持较高,p53–p21通路活动减弱。这一实验表明,维持POLR2A可以帮助细胞抵御本会将其推入永久衰老状态的损伤。
这对健康衰老意味着什么
综合来看,这些发现勾勒出衰老细胞内部的一条新事件链。在应激下,LMO7标记POLR2A以便降解,导致POLR2A蛋白水平下降。其后MDM4的表达减少,使p53积累并激活p21,从而将细胞锁定为衰老状态。研究表明恢复POLR2A可以中断这一链条,因此将LMO7–POLR2A–MDM4–p53–p21通路识别为未来可能延缓组织衰老或清除有害衰老细胞的潜在治疗靶点。尽管任何医学应用还为时尚早,理解这一内部“衰老开关”使科学家在管理细胞老化方式方面又迈进一步。
引用: Lai, C., Fu, W., Liu, J. et al. LMO7-mediated POLR2A degradation promotes cellular senescence through the MDM4/p53/p21 axis. Cell Death Dis 17, 421 (2026). https://doi.org/10.1038/s41419-026-08679-0
关键词: 细胞衰老, 衰老, POLR2A, p53通路, 泛素化