Clear Sky Science · en
LMO7-mediated POLR2A degradation promotes cellular senescence through the MDM4/p53/p21 axis
Why aging cells matter
As we grow older, our cells gradually lose their ability to divide and repair tissues. Many of these worn-out cells do not die; instead, they enter a long‑term “sleep” state called cellular senescence. Senescent cells can weaken organs and drive age‑related diseases by releasing inflammatory factors. This study asks a simple but important question: what makes a healthy cell tip over into this senescent state—and can that switch be controlled?
A key worker in the cell’s copying room
Inside every cell, a large molecular machine called RNA polymerase II reads DNA and helps turn genes on. POLR2A is the largest and most important piece of this machine. Earlier work hinted that POLR2A levels drop in older tissues and in premature‑aging disorders, but no one knew whether this was a side effect of aging or part of the cause. The authors studied human fibroblasts, a common type of support cell in skin and lung, and also looked at tissues from mice of different ages to see how POLR2A changes with time.
Watching cells grow old in the lab
The team used two ways to make cells age in a dish. In “replicative” aging, fibroblasts were simply allowed to divide over and over until they naturally slowed down. In “stress‑induced” aging, young cells were briefly exposed to hydrogen peroxide, which causes oxidative damage. In both models, more and more cells showed classic hallmarks of senescence: they stopped dividing, stained positive for a standard senescence marker, and released inflammatory molecules. Strikingly, as these changes appeared, POLR2A protein levels fell sharply, even though its mRNA did not, indicating that the protein was being destroyed rather than made less often. Similar declines in POLR2A were seen in the brains, hearts and lungs of older mice, linking this process to aging in living animals.
How losing POLR2A flips the aging switch
To test cause and effect, the researchers deliberately lowered POLR2A in young fibroblasts. The cells rapidly took on a senescent identity: they stopped dividing, turned on the senescence marker, and secreted inflammatory signals. At the molecular level, a well‑known safety pathway built around the protein p53 and its partner p21 became highly active. When POLR2A and p53 were knocked down together, the cells largely escaped senescence, showing that POLR2A normally helps prevent aging through this p53–p21 route. Digging deeper, the team found that p53 levels rose not because more p53 was made, but because it was broken down more slowly. A protein called MDM4, which usually helps tag p53 for disposal, dropped when POLR2A fell, leading to more stable p53 and stronger signals to stop cell division.
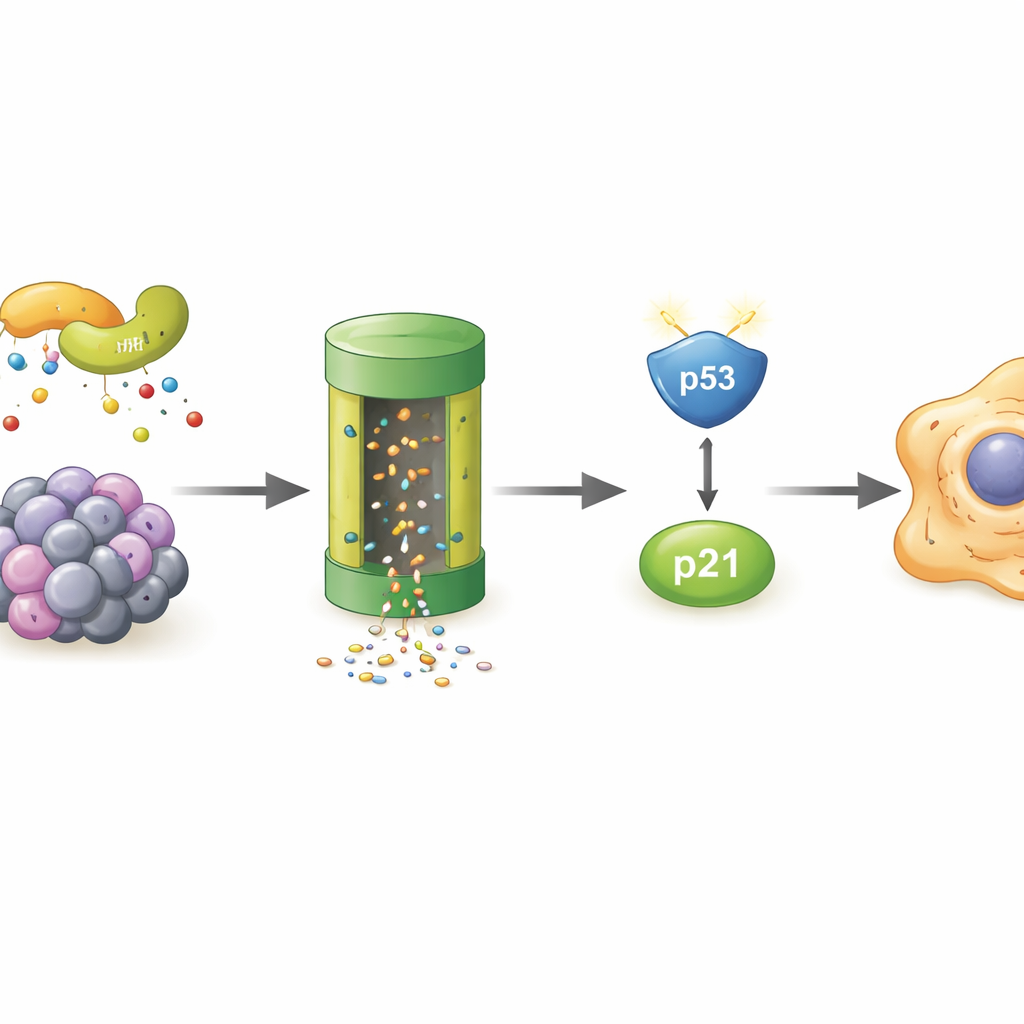
Who destroys POLR2A in aging cells
Since POLR2A itself was disappearing faster in senescent cells, the authors searched for the cellular “trash system” responsible. They showed that blocking the proteasome—the cell’s main protein shredder—rescued POLR2A levels, and that POLR2A carried more ubiquitin tags, molecular flags that target proteins for destruction. A mass‑spectrometry screen singled out LMO7, a scaffold protein that can act as part of an enzyme complex adding these tags. In stressed cells, LMO7 bound more strongly to POLR2A. When LMO7 was removed, POLR2A no longer became heavily tagged with ubiquitin and its levels stayed higher, even under oxidative stress. This pinpointed LMO7 as a central driver of POLR2A loss during cellular aging.
Turning the clock back inside cells
Finally, the researchers asked whether boosting POLR2A could push back against senescence. Using CRISPR activation technology, they gently turned up the cell’s own POLR2A gene rather than adding an artificial copy. Cells with elevated POLR2A were more resistant to both stress‑induced and replicative senescence: fewer became positive for senescence markers, and more kept dividing. At the same time, MDM4 levels stayed higher and the p53–p21 pathway was less active. This experiment suggests that preserving POLR2A can help cells withstand damage that would otherwise push them into a permanent aged state.
What this means for healthy aging
Taken together, the findings outline a new chain of events inside aging cells. Under stress, LMO7 marks POLR2A for destruction, lowering POLR2A protein levels. This, in turn, reduces production of MDM4, allowing p53 to accumulate and activate p21, which locks cells into senescence. By showing that restoring POLR2A can interrupt this chain, the study identifies the LMO7–POLR2A–MDM4–p53–p21 pathway as a potential target for future therapies that aim to delay tissue aging or clear harmful senescent cells. While any medical applications are still far off, understanding this internal “aging switch” brings scientists a step closer to managing how our cells grow old.
Citation: Lai, C., Fu, W., Liu, J. et al. LMO7-mediated POLR2A degradation promotes cellular senescence through the MDM4/p53/p21 axis. Cell Death Dis 17, 421 (2026). https://doi.org/10.1038/s41419-026-08679-0
Keywords: cellular senescence, aging, POLR2A, p53 pathway, ubiquitination