Clear Sky Science · de
LMO7-vermittelte POLR2A‑Degradation fördert zelluläre Seneszenz über die MDM4/p53/p21‑Achse
Warum gealterte Zellen wichtig sind
Mit zunehmendem Alter verlieren unsere Zellen allmählich die Fähigkeit, sich zu teilen und Gewebe zu reparieren. Viele dieser verschlissenen Zellen sterben nicht; stattdessen treten sie in einen langfristigen "Schlaf"-Zustand ein, die sogenannte zelluläre Seneszenz. Seneszente Zellen können Organe schwächen und altersbedingte Krankheiten vorantreiben, indem sie entzündungsfördernde Faktoren freisetzen. Diese Studie stellt eine einfache, aber wichtige Frage: Was bringt eine gesunde Zelle dazu, in diesen seneszenten Zustand umzuschalten — und lässt sich dieser Schalter steuern?
Ein Schlüsselakteur im Kopierraum der Zelle
Im Inneren jeder Zelle liest eine große molekulare Maschine, die RNA‑Polymerase II, die DNA und hilft dabei, Gene zu aktivieren. POLR2A ist das größte und wichtigste Bauteil dieser Maschine. Frühere Arbeiten deuteten an, dass die POLR2A‑Spiegel in älteren Geweben und bei vorzeitigen Alterungsstörungen abnehmen, doch war unklar, ob dies eine Begleiterscheinung des Alterns oder ein ursächlicher Faktor ist. Die Autorinnen und Autoren untersuchten humane Fibroblasten, eine häufige Unterstützerzellenart in Haut und Lunge, und analysierten außerdem Gewebe von Mäusen unterschiedlichen Alters, um zu sehen, wie sich POLR2A im Laufe der Zeit verändert.
Zellen im Labor beim Altwerden beobachten
Das Team verwendete zwei Modelle, um Zellen in der Kultur altern zu lassen. Bei der "replikativen" Alterung wurden Fibroblasten einfach wiederholt teilen gelassen, bis sie sich natürlich verlangsamten. Bei der "stressinduzierten" Alterung wurden junge Zellen kurz Wasserstoffperoxid ausgesetzt, das oxidativen Schaden verursacht. In beiden Modellen zeigten immer mehr Zellen klassische Merkmale der Seneszenz: Sie hörten auf, sich zu teilen, zeigten positiv einen standardmäßigen Seneszenzmarker und setzten entzündliche Moleküle frei. Auffällig war, dass mit dem Auftreten dieser Veränderungen die POLR2A‑Proteinspiegel stark sanken, obwohl die mRNA unverändert blieb — ein Hinweis darauf, dass das Protein zerstört und nicht weniger hergestellt wurde. Ähnliche Rückgänge von POLR2A wurden in Gehirn, Herz und Lunge älterer Mäuse beobachtet und verknüpfen diesen Prozess mit dem Altern in lebenden Organismen.
Wie der Verlust von POLR2A den Alterns‑Schalter umlegt
Um Ursache und Wirkung zu prüfen, reduzierten die Forschenden gezielt POLR2A in jungen Fibroblasten. Die Zellen nahmen rasch eine seneszente Identität an: Sie hörten auf zu proliferieren, zeigten den Seneszenzmarker und sezernierten entzündungsfördernde Signale. Auf molekularer Ebene wurde ein bekannter Sicherheitsweg rund um das Protein p53 und dessen Partner p21 stark aktiviert. Wurden POLR2A und p53 gleichzeitig herunterreguliert, entgingen die Zellen weitgehend der Seneszenz, was zeigt, dass POLR2A gewöhnlich über diesen p53–p21‑Weg das Altern verhindert. Bei genauerer Untersuchung stellte das Team fest, dass die p53‑Spiegel nicht aufgrund gesteigerter Synthese stiegen, sondern weil der Abbau verlangsamt war. Ein Protein namens MDM4, das normalerweise hilft, p53 für den Abbau zu markieren, nahm ab, als POLR2A sank, was zu stabilerem p53 und stärkeren Signalen zum Stopp der Zellteilung führte.
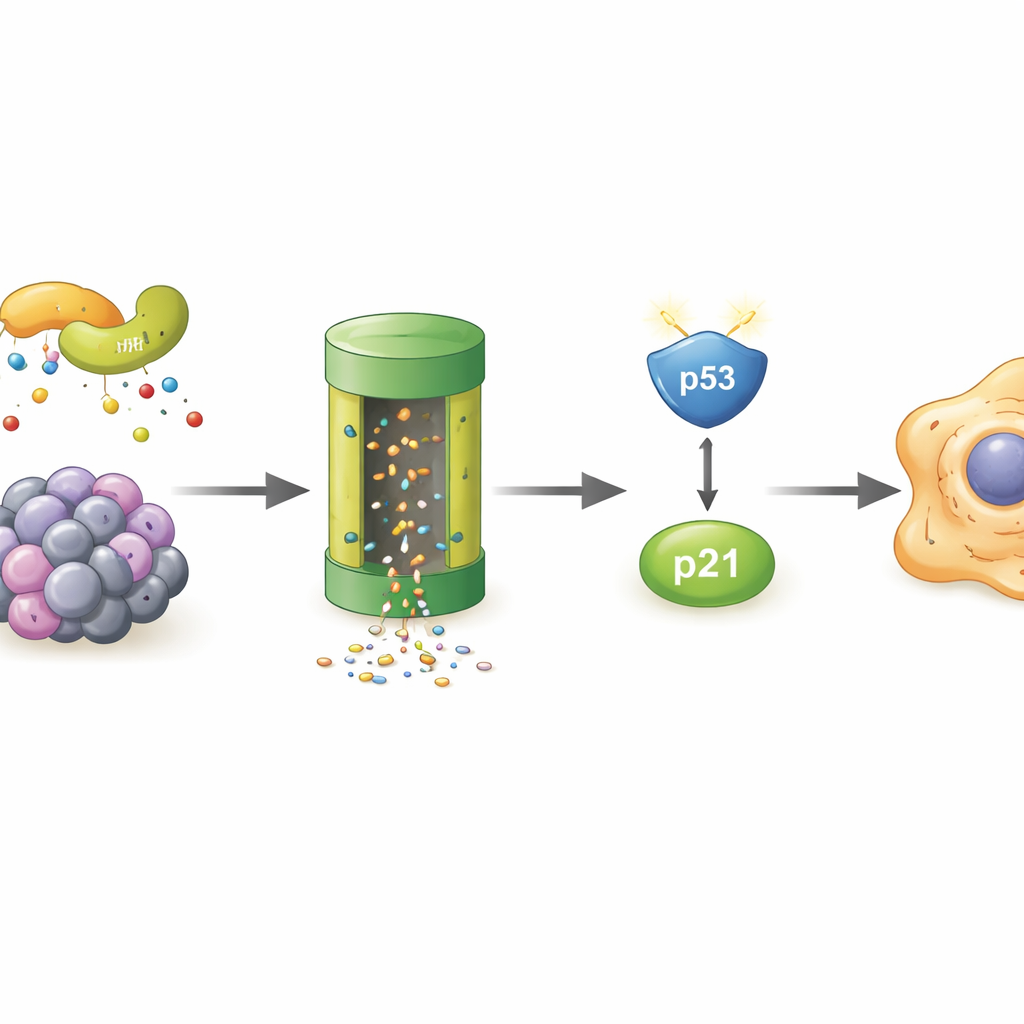
Wer zerstört POLR2A in seneszenten Zellen
Da POLR2A in seneszenten Zellen selbst schneller verschwand, suchten die Autorinnen und Autoren nach dem zellulären "Müllsystem", das dafür verantwortlich ist. Sie zeigten, dass das Blockieren des Proteasoms — dem hauptsächlichen Protein‑Shredder der Zelle — POLR2A‑Spiegel rettete und dass POLR2A mehr Ubiquitin‑Markierungen trug, molekulare Flaggen, die Proteine für den Abbau kennzeichnen. Eine Massenspektrometrie‑Suche identifizierte LMO7, ein Gerüstprotein, das als Teil eines Enzymkomplexes wirken kann, der diese Markierungen anbringt. In gestressten Zellen band LMO7 stärker an POLR2A. Wurde LMO7 entfernt, wurde POLR2A nicht mehr stark mit Ubiquitin markiert und seine Spiegel blieben auch unter oxidativem Stress höher. Das macht LMO7 zu einem zentralen Treiber des POLR2A‑Verlusts während der zellulären Alterung.
Die Uhr in Zellen zurückdrehen
Schließlich fragten die Forschenden, ob das Anheben von POLR2A der Seneszenz entgegenwirken kann. Mittels CRISPR‑Aktivierungstechnologie erhöhten sie schonend das eigene POLR2A‑Gen der Zelle, anstatt eine künstliche Kopie hinzuzufügen. Zellen mit erhöhtem POLR2A widerstanden sowohl stressinduzierter als auch replikativer Seneszenz besser: Weniger Zellen wurden seneszenzmarker‑positiv, und mehr teilten sich weiterhin. Gleichzeitig blieben die MDM4‑Spiegel höher und der p53–p21‑Weg war weniger aktiv. Dieses Experiment legt nahe, dass der Erhalt von POLR2A Zellen dabei helfen kann, Schäden zu überstehen, die sie sonst in einen dauerhaften gealterten Zustand treiben würden.
Was das für gesundes Altern bedeutet
Insgesamt skizzieren die Befunde eine neue Kette von Ereignissen in alternden Zellen. Unter Stress markiert LMO7 POLR2A für den Abbau, wodurch die POLR2A‑Proteinspiegel sinken. Dies verringert wiederum die Produktion von MDM4, sodass p53 akkumuliert und p21 aktiviert wird, was Zellen in die Seneszenz einschließt. Indem gezeigt wird, dass die Wiederherstellung von POLR2A diese Kette unterbrechen kann, identifiziert die Studie die LMO7–POLR2A–MDM4–p53–p21‑Achse als potenzielles Ziel für künftige Therapien, die darauf abzielen, das Gewebealter zu verzögern oder schädliche seneszente Zellen zu entfernen. Während medizinische Anwendungen noch in weiter Ferne liegen, bringt das Verständnis dieses inneren "Altersschalters" die Wissenschaft einen Schritt näher daran, zu steuern, wie unsere Zellen alt werden.
Zitation: Lai, C., Fu, W., Liu, J. et al. LMO7-mediated POLR2A degradation promotes cellular senescence through the MDM4/p53/p21 axis. Cell Death Dis 17, 421 (2026). https://doi.org/10.1038/s41419-026-08679-0
Schlüsselwörter: zelluläre Seneszenz, Altern, POLR2A, p53‑Signalweg, Ubiquitinierung