Clear Sky Science · sv
LMO7-medierad nedbrytning av POLR2A främjar cellulär senescens via MDM4/p53/p21-axeln
Varför åldrande celler är viktiga
När vi blir äldre förlorar våra celler gradvis förmågan att dela sig och reparera vävnader. Många av dessa utslitna celler dör inte; i stället går de in i ett långvarigt ”sömnläge” som kallas cellulär senescens. Senescenta celler kan försvaga organ och driva åldersrelaterade sjukdomar genom att utsöndra inflammatoriska faktorer. Denna studie ställer en enkel men viktig fråga: vad får en frisk cell att gå över gränsen till detta senescenta tillstånd — och kan den omkopplaren kontrolleras?
En nyckelspelare i cellens kopieringsverkstad
Inuti varje cell läser en stor molekylär maskin kallad RNA-polymeras II av DNA och hjälper till att slå på gener. POLR2A är den största och viktigaste delen av denna maskin. Tidigare arbete antydde att POLR2A-nivåer sjunker i äldre vävnader och vid förtida åldrande, men ingen visste om detta var en bieffekt av åldrande eller en del av orsaken. Författarna studerade humana fibroblaster, en vanlig typ av stödjande cell i hud och lunga, och undersökte även vävnader från möss i olika åldrar för att se hur POLR2A förändras över tid.
Att iaktta celler åldras i laboratoriet
Teamet använde två sätt för att åstadkomma åldrande i en odlingsskål. Vid ”replikativt” åldrande tilläts fibroblaster helt enkelt dela sig om och om igen tills de naturligt saktade ner. Vid ”stressinducerat” åldrande utsattes unga celler kort för väteperoxid, vilket orsakar oxidativ skada. I båda modellerna visade fler och fler celler klassiska kännetecken för senescens: de slutade dela sig, färgades positiva för en standardmarkör för senescens och utsöndrade inflammatoriska molekyler. Slående nog föll POLR2A-proteinnivåerna kraftigt i samband med dessa förändringar, även om dess mRNA inte minskade, vilket indikerar att proteinet förstördes snarare än att det producerades mindre. Liknande minskningar av POLR2A sågs i hjärnor, hjärtan och lungor hos äldre möss, vilket kopplar denna process till åldrande i levande djur.
Hur förlust av POLR2A slår om åldersströmbrytaren
För att testa orsak och verkan sänkte forskarna medvetet POLR2A i unga fibroblaster. Cellerna antog snabbt en senescent identitet: de slutade dela sig, uttryckte senescensmarkören och sekretorerade inflammatoriska signaler. På molekylär nivå blev en välkänd säkerhetsväg uppbyggd kring proteinet p53 och dess partner p21 mycket aktiv. När POLR2A och p53 släcktes ned samtidigt undkom cellerna till stor del senescens, vilket visar att POLR2A normalt hjälper till att förhindra åldrande via denna p53–p21-väg. Vid närmare undersökning fann teamet att p53-nivåerna ökade inte därför att mer p53 syntetiserades, utan för att det bröts ner långsammare. Ett protein kallat MDM4, som normalt hjälper till att märka p53 för nedbrytning, sjönk när POLR2A föll, vilket ledde till stabilare p53 och starkare signaler att stoppa celldelning.
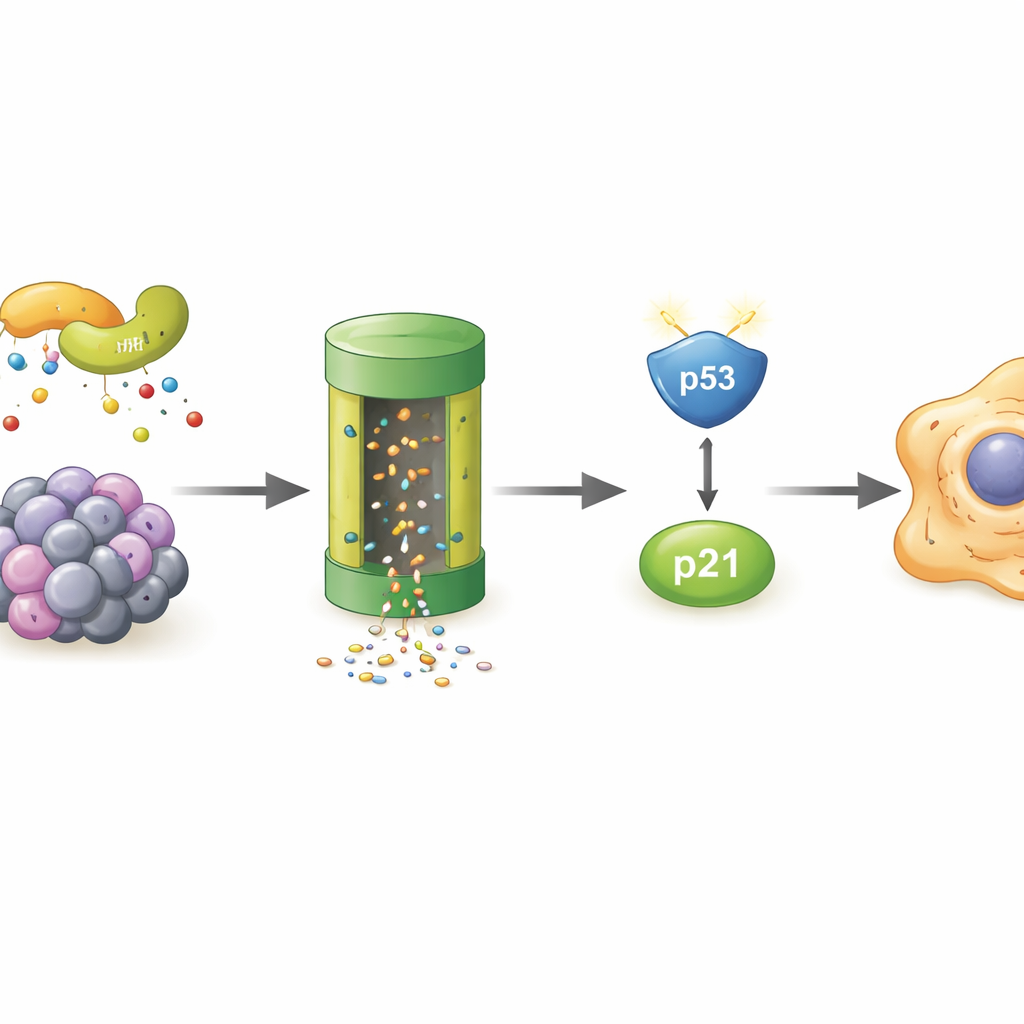
Vem förstör POLR2A i åldrande celler
Eftersom POLR2A själv försvann snabbare i senescenta celler sökte författarna efter det cellulära ”sop‑system” som låg bakom. De visade att blockering av proteasomen — cellens huvudsakliga proteinförstöringsmaskin — räddade POLR2A‑nivåerna, och att POLR2A bar fler ubiquitin‑märken, molekylära flaggor som riktar proteiner för nedbrytning. En masspektrometriscreen pekade ut LMO7, ett scaffoldprotein som kan ingå i ett enzymkomplex som lägger till dessa märken. I stressade celler band LMO7 starkare till POLR2A. När LMO7 avlägsnades blev POLR2A inte längre tungt märkt med ubiquitin och dess nivåer hölls högre, även under oxidativ stress. Detta pekar på LMO7 som en central drivkraft bakom POLR2A‑förlust under cellulärt åldrande.
Att vrida tillbaka klockan inuti celler
Slutligen frågade forskarna om ökade POLR2A-nivåer kunde motverka senescens. Med CRISPR‑aktiveringsteknik ökade de försiktigt cellens eget POLR2A‑genuttryck i stället för att tillsätta en konstgjord kopia. Celler med förhöjd POLR2A var mer motståndskraftiga mot både stressinducerad och replikativ senescens: färre blev positiva för senescensmarkörer och fler fortsatte dela sig. Samtidigt hölls MDM4‑nivåerna högre och p53–p21‑banan var mindre aktiv. Denna experimentella observation tyder på att bevarande av POLR2A kan hjälpa celler att stå emot skada som annars skulle skjuta dem in i ett permanent åldrat tillstånd.
Vad detta betyder för ett hälsosamt åldrande
Tillsammans skisserar resultaten en ny händelsekedja inne i åldrande celler. Under stress markerar LMO7 POLR2A för nedbrytning och sänker därmed POLR2A‑proteinnivåerna. Detta minskar i sin tur produktionen av MDM4, vilket tillåter p53 att ackumuleras och aktivera p21, som låser celler i senescens. Genom att visa att återställning av POLR2A kan avbryta denna kedja identifierar studien LMO7–POLR2A–MDM4–p53–p21‑vägen som ett potentiellt mål för framtida terapier som syftar till att fördröja vävnadsåldrande eller rensa skadliga senescenta celler. Medan eventuella medicinska tillämpningar ännu ligger långt fram i tiden, för oss en bättre förståelse av denna interna ”åldersomkopplare” forskare ett steg närmare att kunna hantera hur våra celler åldras.
Citering: Lai, C., Fu, W., Liu, J. et al. LMO7-mediated POLR2A degradation promotes cellular senescence through the MDM4/p53/p21 axis. Cell Death Dis 17, 421 (2026). https://doi.org/10.1038/s41419-026-08679-0
Nyckelord: cellulär senescens, åldrande, POLR2A, p53-vägen, ubiquitinering