Clear Sky Science · pl
Degradacja POLR2A zależna od LMO7 promuje starzenie komórkowe poprzez oś MDM4/p53/p21
Dlaczego starejące komórki mają znaczenie
W miarę jak się starzejemy, nasze komórki stopniowo tracą zdolność do podziału i naprawy tkanek. Wiele z tych zużytych komórek nie ginie; zamiast tego wchodzi w długotrwały stan „uśpienia” zwany starzeniem komórkowym (senescencją). Komórki senescentne osłabiają funkcję narządów i napędzają choroby związane z wiekiem przez uwalnianie czynników zapalnych. W tym badaniu postawiono proste, ale istotne pytanie: co sprawia, że zdrowa komórka przechodzi w ten stan senescencji — i czy można ten przełącznik kontrolować?
Kluczowy operator w komórkowym dziale kopiowania
W każdej komórce duża maszyna molekularna zwana polimerazą RNA II odczytuje DNA i pomaga aktywować geny. POLR2A jest największym i najważniejszym elementem tej maszyny. Wcześniejsze prace sugerowały, że poziomy POLR2A spadają w starszych tkankach i w chorobach przyspieszonego starzenia, ale nie wiadomo było, czy to efekt uboczny starzenia, czy jego przyczyna. Autorzy badali ludzkie fibroblasty — powszechny typ komórek podporowych skóry i płuc — oraz analizowali tkanki myszy w różnym wieku, aby zobaczyć, jak POLR2A zmienia się w czasie.
Obserwowanie starzenia komórek w laboratorium
Zespół zastosował dwa sposoby wywoływania starzenia komórek na płytce. W „starzeniu replikacyjnym” fibroblasty były po prostu pozwalane na wielokrotne podziały, aż naturalnie zwolniły. W „starzeniu indukowanym stresem” młode komórki krótko eksponowano na nadtlenek wodoru, który powoduje uszkodzenia oksydacyjne. W obu modelach coraz więcej komórek wykazywało klasyczne cechy senescencji: przestawały się dzielić, barwiły się dodatnio na standardowy marker senescencji i wydzielały czynniki zapalne. Co uderzające, wraz z tymi zmianami poziom białka POLR2A gwałtownie spadał, mimo że jego mRNA nie ulegało znacznemu obniżeniu, co wskazuje, że białko było szybciej degradowane, a nie mniej wytwarzane. Podobne spadki POLR2A zaobserwowano w mózgach, sercach i płucach starszych myszy, łącząc ten proces ze starzeniem w organizmach żywych.
Jak utrata POLR2A przełącza komórkę na tryb starzenia
Aby sprawdzić związek przyczynowo‑skutkowy, badacze celowo obniżyli poziom POLR2A w młodych fibroblastach. Komórki szybko przyjęły tożsamość senescentną: przestały się dzielić, włączyły marker senescencji i zaczęły wydzielać sygnały zapalne. Na poziomie molekularnym silnie aktywował się dobrze znany szlak zabezpieczający oparty na białku p53 i jego partnerze p21. Gdy jednocześnie obniżono POLR2A i p53, komórki w dużej mierze uniknęły senescencji, co wykazało, że POLR2A normalnie pomaga zapobiegać starzeniu poprzez tę drogę p53–p21. Wnikliwsze analizy pokazały, że poziom p53 wzrastał nie dlatego, że powstawało go więcej, lecz dlatego, że był wolniej rozkładany. Białko MDM4, które zwykle pomaga oznaczać p53 do utylizacji, spadało po utracie POLR2A, co prowadziło do stabilniejszego p53 i silniejszych sygnałów zatrzymujących podział komórkowy.
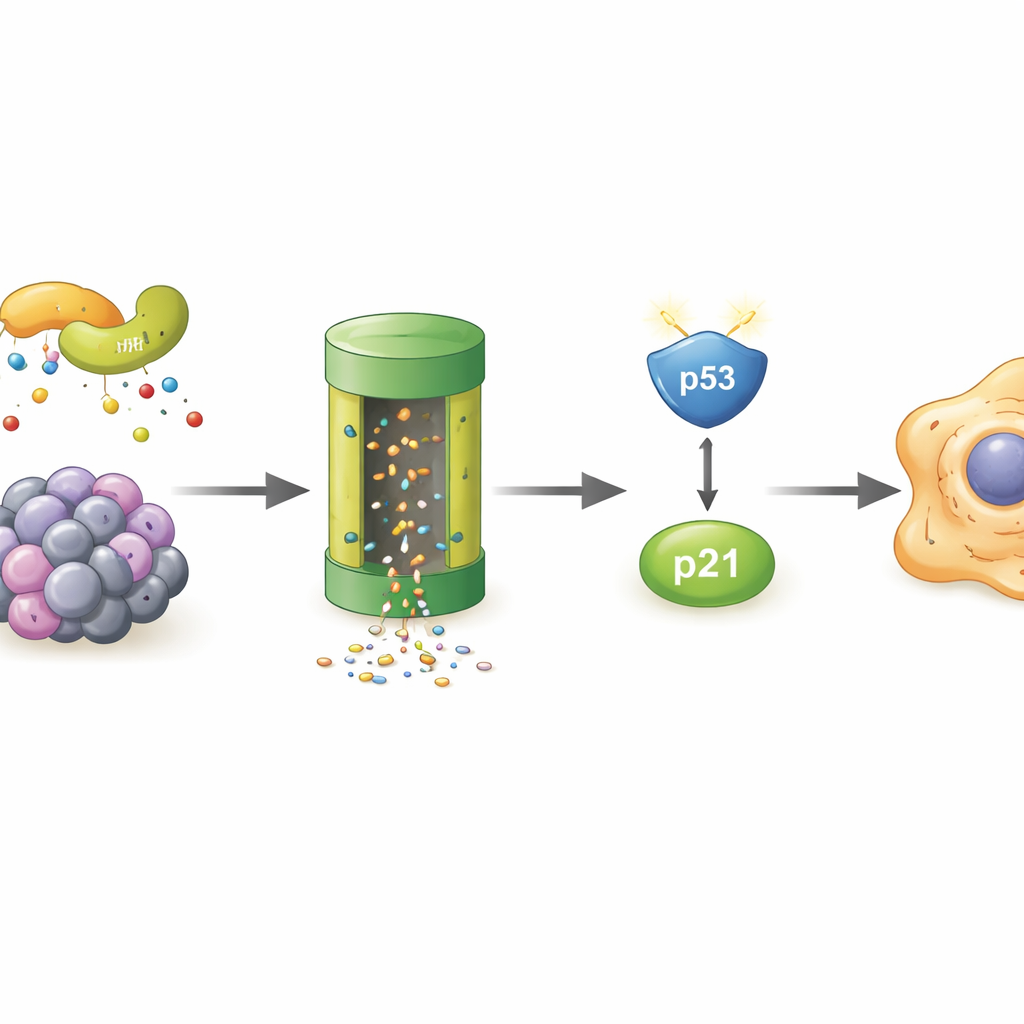
Kto niszczy POLR2A w starzejących się komórkach
Skoro samo POLR2A znikało szybciej w komórkach senescentnych, autorzy poszukiwali systemu „utylizacji” odpowiedzialnego za ten proces. Wykazali, że blokada proteasomu — głównego komórkowego „niszczarki” białek — ratowała poziomy POLR2A, oraz że POLR2A nosiło więcej znaczników ubikwityny, molekularnych flag kierujących białka do degradacji. Skanowanie za pomocą spektrometrii mas wyłoniło LMO7, białko‑rusztowanie, które może działać jako część kompleksu enzymatycznego dodającego te tagi. W komórkach poddanych stresowi LMO7 wiązało się silniej z POLR2A. Gdy usunięto LMO7, POLR2A nie było już intensywnie oznaczane ubikwityną i jego poziomy utrzymywały się wyżej, nawet przy stresie oksydacyjnym. To wskazuje na LMO7 jako centralnego sprawcę utraty POLR2A podczas starzenia komórkowego.
Przywracanie zegara wewnątrz komórek
Na koniec badacze zapytali, czy zwiększenie poziomu POLR2A może przeciwdziałać senescencji. Z użyciem technologii aktywacji CRISPR delikatnie wzmocnili ekspresję własnego genu POLR2A, zamiast wprowadzać sztuczną kopię. Komórki z podwyższonym POLR2A były bardziej odporne na senescencję indukowaną stresem i replikacyjną: mniej z nich było dodatnich na markery senescencji, a więcej nadal się dzieliło. Jednocześnie utrzymywały się wyższe poziomy MDM4, a szlak p53–p21 był mniej aktywny. Ten eksperyment sugeruje, że zachowanie poziomu POLR2A może pomóc komórkom przetrwać uszkodzenia, które w przeciwnym razie pchnęłyby je w stan trwałego starzenia.
Co to oznacza dla zdrowego starzenia
W sumie wyniki opisują nowy łańcuch zdarzeń wewnątrz starzejących się komórek. Pod wpływem stresu LMO7 oznacza POLR2A do degradacji, obniżając poziomy białka POLR2A. To z kolei zmniejsza produkcję MDM4, pozwalając na akumulację p53 i aktywację p21, które blokują komórki w stanie senescencji. Pokazując, że przywrócenie POLR2A może przerwać ten łańcuch, badanie identyfikuje szlak LMO7–POLR2A–MDM4–p53–p21 jako potencjalny cel przyszłych terapii mających opóźniać starzenie tkanek lub usuwać szkodliwe komórki senescentne. Choć zastosowania medyczne są wciąż odległe, zrozumienie tego wewnętrznego „przełącznika starzenia” przybliża naukowców do kontroli procesu, w którym nasze komórki się starzeją.
Cytowanie: Lai, C., Fu, W., Liu, J. et al. LMO7-mediated POLR2A degradation promotes cellular senescence through the MDM4/p53/p21 axis. Cell Death Dis 17, 421 (2026). https://doi.org/10.1038/s41419-026-08679-0
Słowa kluczowe: starzenie komórkowe, starzenie, POLR2A, szlak p53, ubikwitynacja